
Proteomic Alteration in the Progression of Multiple Myeloma: A Comprehensive Review
 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
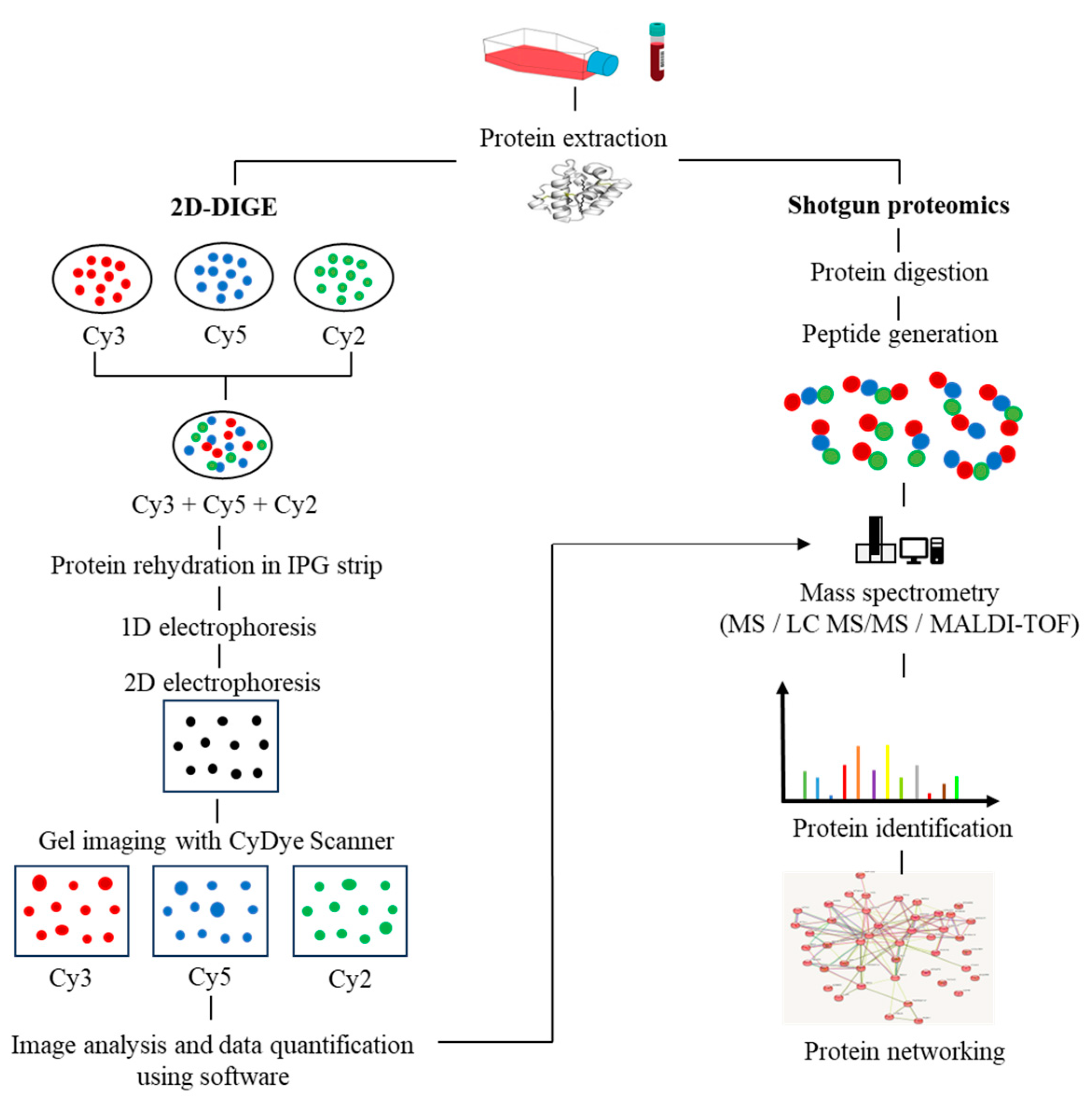
2. Proteomics Breakthroughs in Protein Identification
3. Post-Translational Modifications in MM
4. Proteomics Alteration in MM
4.1. Monoclonal Gammopathy of Undetermined Significance (MGUS)
4.2. Smoldering Multiple Myeloma (SMM)
4.3. Multiple Myeloma (MM)
4.4. Myeloma Bone Disease
4.5. Relapse/Refractory MM
4.6. Extramedullary MM
5. Investigating Biomarkers in Drug-Resistant MM
6. Therapeutic Agents to Target Protein Signatures
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hari, P.; Mateos, M.V.; Abonour, R.; Knop, S.; Bensinger, W.; Ludwig, H.; Song, K.; Hajek, R.; Moreau, P.; Siegel, D.S.; et al. Efficacy and safety of carfilzomib regimens in multiple myeloma patients relapsing after autologous stem cell transplant: ASPIRE and ENDEAVOR outcomes. Leukemia 2017, 31, 2630–2641. [Google Scholar] [CrossRef] [PubMed]
- Tsang, M.; Le, M.; Ghazawi, F.M.; Cyr, J.; Alakel, A.; Rahme, E.; Lagacé, F.; Netchiporouk, E.; Moreau, L.; Zubarev, A.; et al. Multiple myeloma epidemiology and patient geographic distribution in Canada: A population study. Cancer 2019, 125, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2022, 97, 1086–1107. [Google Scholar] [CrossRef] [PubMed]
- Prideaux, S.M.; Conway O’Brien, E.; Chevassut, T.J. The genetic architecture of multiple myeloma. Adv. Hematol. 2014, 2014, 864058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Kawano, Y.; Bianchi, G.; Roccaro, A.M.; Ghobrial, I.M. Cell autonomous and microenvironmental regulation of tumor progression in precursor states of multiple myeloma. Curr. Opin. Hematol. 2016, 23, 426–433. [Google Scholar] [CrossRef]
- Bezieau, S.; Devilder, M.C.; Avet-Loiseau, H.; Mellerin, M.P.; Puthier, D.; Pennarun, E.; Rapp, M.J.; Harousseau, J.L.; Moisan, J.P.; Bataille, R. High incidence of N and K-Ras activating mutations in multiple myeloma and primary plasma cell leukemia at diagnosis. Hum. Mutat. 2001, 18, 212–224. [Google Scholar] [CrossRef]
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef]
- Ismail, N.H.; Mussa, A.; Zakaria, N.A.; Al-Khreisat, M.J.; Zahidin, M.A.; Ramli, N.N.; Mohammad, S.; Hassan, R.; Mohd Noor, N.H.; Iberahim, S.; et al. The Role of Epigenetics in the Development and Progression of Multiple Myeloma. Biomedicines 2022, 10, 2767. [Google Scholar] [CrossRef]
- Zhan, F.; Hardin, J.; Kordsmeier, B.; Bumm, K.; Zheng, M.; Tian, E.; Sanderson, R.; Yang, Y.; Wilson, C.; Zangari, M.; et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood 2002, 99, 1745–1757. [Google Scholar] [CrossRef]
- Noone, A.-M.; Cronin, K.A.; Altekruse, S.F.; Howlader, N.; Lewis, D.R.; Petkov, V.I.; Penberthy, L. Cancer Incidence and Survival Trends by Subtype Using Data from the Surveillance Epidemiology and End Results Program, 1992–2013. Cancer Epidemiol. Biomarkers Prev. 2017, 26, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia 2013, 28, 1122–1128. [Google Scholar] [CrossRef] [Green Version]
- Mikhael, J.; Bhutani, M.; Cole, C.E. Multiple Myeloma for the Primary Care Provider: A Practical Review to Promote Earlier Diagnosis Among Diverse Populations. Am. J. Med. 2022, 136, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Nooka, A.K.; Kastritis, E.; Dimopoulos, M.A.; Lonial, S. Treatment options for relapsed and refractory multiple myeloma. Blood 2015, 125, 3085–3099. [Google Scholar] [CrossRef] [PubMed]
- Cejalvo, M.J.; de la Rubia, J. Which therapies will move to the front line for multiple myeloma? Expert Rev. Hematol. 2017, 10, 383–392. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: Every year a new standard? Hematol. Oncol. 2019, 37 (Suppl. S1), 62–65. [Google Scholar] [CrossRef] [Green Version]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.G.; Sokolowska, I.; Ngounou Wetie, A.G.; Channaveerappa, D.; Dupree, E.J.; Jayathirtha, M.; Aslebagh, R.; Wormwood, K.L.; Darie, C.C. Mass Spectrometry for Proteomics-Based Investigation. In Advancements of Mass Spectrometry in Biomedical Research; Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1140, pp. 1–26. [Google Scholar] [CrossRef]
- Schirle, M.; Bantscheff, M.; Kuster, B. Mass Spectrometry-Based Proteomics in Preclinical Drug Discovery. Chem. Biol. 2012, 19, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Crutchfield, C.A.; Thomas, S.N.; Sokoll, L.J.; Chan, D.W. Advances in mass spectrometry-based clinical biomarker discovery. Clin. Proteom. 2016, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Sperling, K. From proteomics to genomics. Electrophoresis 2001, 22, 2835–2837. [Google Scholar] [CrossRef]
- Loo, J.A.; DeJohn, D.E.; Du, P.; Stevenson, T.I.; Ogorzalek Loo, R.R. Application of mass spectrometry for target identification and characterization. Med. Res. Rev. 1999, 19, 307–319. [Google Scholar] [CrossRef]
- Yates, J.R., 3rd; McCormack, A.L.; Schieltz, D.; Carmack, E.; Link, A. Direct analysis of protein mixtures by tandem mass spectrometry. J. Protein Chem. 1997, 16, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Marouga, R.; David, S.; Hawkins, E. The development of the DIGE system: 2D fluorescence difference gel analysis technology. Anal. Bioanal. Chem. 2005, 382, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Aebersold, R.; Schwikowski, B. ProbID: A probabilistic algorithm to identify peptides through sequence database searching using tandem mass spectral data. Proteomics 2002, 2, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.-L.; Chen, S.-F. A Brief Review of Bioinformatics Tools for Glycosylation Analysis by Mass Spectrometry. Mass Spectrom. 2017, 6, S0064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köcher, T.; Superti-Furga, G. Mass spectrometry–based functional proteomics: From molecular machines to protein networks. Nat. Methods 2007, 4, 807–815. [Google Scholar] [CrossRef]
- Guang, M.H.Z.; Kavanagh, E.L.; Dunne, L.P.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.Y.; Hanley, C.; Bianchi, G.; et al. Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis. Cancers 2019, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Van Puyvelde, B.; Daled, S.; Willems, S.; Gabriels, R.; Gonzalez de Peredo, A.; Chaoui, K.; Mouton-Barbosa, E.; Bouyssié, D.; Boonen, K.; Hughes, C.J.; et al. A comprehensive LFQ benchmark dataset on modern day acquisition strategies in proteomics. Sci. Data 2022, 9, 126. [Google Scholar] [CrossRef]
- Dowell, J.A.; Wright, L.J.; Armstrong, E.A.; Denu, J.M. Benchmarking Quantitative Performance in Label-Free Proteomics. ACS Omega 2021, 6, 2494–2504. [Google Scholar] [CrossRef]
- O’Rourke, M.B.; Sahni, S.; Samra, J.; Mittal, A.; Molloy, M.P. Data independent acquisition of plasma biomarkers of response to neoadjuvant chemotherapy in pancreatic ductal adenocarcinoma. J. Proteom. 2020, 231, 103998. [Google Scholar] [CrossRef]
- Muntel, J.; Kirkpatrick, J.; Bruderer, R.; Huang, T.; Vitek, O.; Ori, A.; Reiter, L. Comparison of Protein Quantification in a Complex Background by DIA and TMT Workflows with Fixed Instrument Time. J. Proteome Res. 2019, 18, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, L.; Cummins, E.; Samudio, I.; Kislinger, T. Cell-surface proteomics for the identification of novel therapeutic targets in cancer. Expert Rev. Proteom. 2018, 15, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Elschenbroich, S.; Kim, Y.; Medin, J.A.; Kislinger, T. Isolation of cell surface proteins for mass spectrometry-based proteomics. Expert Rev. Proteom. 2010, 7, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Accardi, F.; Marchica, V.; Dalla Palma, B.; Storti, P.; Toscani, D.; Vicario, E.; Malavasi, F. Novel targets for the treatment of relapsing multiple myeloma. Expert Rev. Hematol. 2019, 12, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.; Dalla Palma, B.; Giuliani, N. CD38 Expression by Myeloma Cells and Its Role in the Context of Bone Marrow Microenvironment: Modulation by Therapeutic Agents. Cells 2019, 8, 1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.-H.; Lee, H.-J.; Vo, M.-C.; Kim, H.-J.; Lee, J.-J. Immunotherapy for the treatment of multiple myeloma. Crit. Rev. Oncol./Hematol. 2017, 111, 87–93. [Google Scholar] [CrossRef]
- Ferguson, I.D.; Patiño-Escobar, B.; Tuomivaara, S.T.; Lin, Y.-H.T.; Nix, M.A.; Leung, K.K.; Kasap, C.; Ramos, E.; Vasquez, W.N.; Talbot, A.; et al. The surfaceome of multiple myeloma cells suggests potential immunotherapeutic strategies and protein markers of drug resistance. Nat. Commun. 2022, 13, 4121. [Google Scholar] [CrossRef] [PubMed]
- Patiño-Escobar, B.; Ferguson, I.D.; Wiita, A.P. Unraveling the surface proteomic profile of multiple myeloma to reveal new immunotherapeutic targets and markers of drug resistance. Cell Stress 2022, 6, 89–92. [Google Scholar] [CrossRef]
- Kelly, R.T. Single-cell Proteomics: Progress and Prospects. Mol. Cell. Proteom. 2020, 19, 1739–1748. [Google Scholar] [CrossRef]
- Chen, M.; Jiang, J.; Hou, J. Single-cell technologies in multiple myeloma: New insights into disease pathogenesis and translational implications. Biomark. Res. 2023, 11, 55. [Google Scholar] [CrossRef]
- Becht, E.; Tolstrup, D.; Dutertre, C.-A.; Morawski, P.A.; Campbell, D.J.; Ginhoux, F.; Newell, E.W.; Gottardo, R.; Headley, M.B.; Lee, L.; et al. High-throughput single-cell quantification of hundreds of proteins using conventional flow cytometry and machine learning. Sci. Adv. 2021, 7, eabg0505. [Google Scholar] [CrossRef] [PubMed]
- Erfanian, N.; Heydari, A.A.; Ianez, P.; Derakhshani, A.; Ghasemigol, M.; Farahpour, M.; Nasseri, S.; Safarpour, H.; Sahebkar, A.J.b. Deep learning applications in single-cell omics data analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Barabási, A.-L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Zybailov, B.L.; Byrd, A.K.; Glazko, G.V.; Rahmatallah, Y.; Raney, K.D. Protein-protein interaction analysis for functional characterization of helicases. Methods 2016, 108, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Ge, F.; Xiao, C.-L.; Yin, X.-F.; Lu, C.-H.; Zeng, H.-L.; He, Q.-Y. Phosphoproteomic analysis of primary human multiple myeloma cells. J. Proteom. 2010, 73, 1381–1390. [Google Scholar] [CrossRef]
- Ge, F.; Xiao, C.-L.; Bi, L.-J.; Tao, S.-C.; Xiong, S.; Yin, X.-F.; Li, L.-P.; Lu, C.-H.; Jia, H.-T.; He, Q.-Y. Quantitative phosphoproteomics of proteasome inhibition in multiple myeloma cells. PLoS ONE 2010, 5, e13095. [Google Scholar] [CrossRef]
- Pollett, J.B.; Trudel, S.; Stern, D.; Li, Z.H.; Stewart, A.K. Overexpression of the myeloma-associated oncogene fibroblast growth factor receptor 3 confers dexamethasone resistance. Blood 2002, 100, 3819–3821. [Google Scholar] [CrossRef]
- Bondt, A.; Rombouts, Y.; Selman, M.H.; Hensbergen, P.J.; Reiding, K.R.; Hazes, J.M.; Dolhain, R.J.; Wuhrer, M. Immunoglobulin G (IgG) Fab glycosylation analysis using a new mass spectrometric high-throughput profiling method reveals pregnancy-associated changes. Mol. Cell. Proteom. 2014, 13, 3029–3039. [Google Scholar] [CrossRef]
- Van de Bovenkamp, F.S.; Hafkenscheid, L.; Rispens, T.; Rombouts, Y. The Emerging Importance of IgG Fab Glycosylation in Immunity. J. Immunol. 2016, 196, 1435–1441. [Google Scholar] [CrossRef] [Green Version]
- Gornik, O.; Pavić, T.; Lauc, G. Alternative glycosylation modulates function of IgG and other proteins—Implications on evolution and disease. Biochim. Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 1318–1326. [Google Scholar] [CrossRef]
- Murata, S.; Yashiroda, H.; Tanaka, K. Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol. 2009, 10, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting Proteostasis for Disease Intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Zhu, P.; Wang, J.; Pascual, G.; Ohgi, K.A.; Lozach, J.; Glass, C.K.; Rosenfeld, M.G. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol. Cell 2008, 29, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, S.; Chahwan, R.; Nepal, R.M.; Frieder, D.; Panier, S.; Roa, S.; Zaheen, A.; Durocher, D.; Scharff, M.D.; Martin, A. The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proc. Natl. Acad. Sci. USA 2009, 107, 809–814. [Google Scholar] [CrossRef]
- Zhan, F.; Barlogie, B.; Arzoumanian, V.; Huang, Y.; Williams, D.R.; Hollmig, K.; Pineda-Roman, M.; Tricot, G.; van Rhee, F.; Zangari, M.; et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood 2006, 109, 1692–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chng, W.J.; Kumar, S.; VanWier, S.; Ahmann, G.; Price-Troska, T.; Henderson, K.; Chung, T.H.; Kim, S.; Mulligan, G.; Bryant, B.; et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res 2007, 67, 2982–2989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagani, Z.; Wiederschain, D.; Loo, A.; He, D.; Mosher, R.; Fordjour, P.; Monahan, J.; Morrissey, M.; Yao, Y.-M.; Lengauer, C.; et al. The Polycomb group protein Bmi-1 Is essential for the growth of multiple myeloma cells. Cancer Res 2010, 70, 5528–5538. [Google Scholar] [CrossRef] [Green Version]
- Roy, P.; Sarkar, U.A.; Basak, S. The NF-κB activating pathways in multiple myeloma. Biomedicines 2018, 6, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Andel, H.; Kocemba, K.A.; de Haan-Kramer, A.; Mellink, C.H.; Piwowar, M.; Broijl, A.; Van Duin, M.; Sonneveld, P.; Maurice, M.; Kersten, M.J.; et al. Loss of CYLD expression unleashes Wnt signaling in multiple myeloma and is associated with aggressive disease. Oncogene 2016, 36, 2105–2115. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.-J.; Van Wier, S.; Tiedemann, R.; Shi, C.-X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Jensen, O.N. Modification-specific proteomics: Strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 2009, 9, 4632–4641. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and Functional Diversity of Lysine Acetylation Revealed by a Proteomics Survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef]
- Zhang, J.; Sprung, R.; Pei, J.; Tan, X.; Kim, S.; Zhu, H.; Liu, C.-F.; Grishin, N.V.; Zhao, Y. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteom. 2009, 8, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.-E.; Mittler, G.; Mann, M. Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat. Methods 2004, 1, 119–126. [Google Scholar] [CrossRef]
- Zhan, X.; Desiderio, D.M. Nitroproteins from a human pituitary adenoma tissue discovered with a nitrotyrosine affinity column and tandem mass spectrometry. Anal. Biochem. 2006, 354, 279–289. [Google Scholar] [CrossRef]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.; Podtelejnikov, A.V.; Blagoev, B.; Bustelo, X.R.; Mann, M.; Lodish, H.F. Analysis of receptor signaling pathways by mass spectrometry: Identification of Vav-2 as a substrate of the epidermal and platelet-derived growth factor receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kruijsbergen, I.; Mulder, M.P.C.; Uckelmann, M.; van Welsem, T.; de Widt, J.; Spanjaard, A.; Jacobs, H.; El Oualid, F.; Ovaa, H.; van Leeuwen, F. Strategy for Development of Site-Specific Ubiquitin Antibodies. Front. Chem. 2020, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vocadlo, D.J.; Hang, H.C.; Kim, E.J.; Hanover, J.A.; Bertozzi, C.R. A chemical approach for identifying O -GlcNAc-modified proteins in cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9116–9121. [Google Scholar] [CrossRef]
- Baskin, J.M.; Prescher, J.A.; Laughlin, S.T.; Agard, N.J.; Chang, P.V.; Miller, I.A.; Lo, A.; Codelli, J.A.; Bertozzi, C.R. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. USA 2007, 104, 16793–16797. [Google Scholar] [CrossRef]
- Kho, Y.; Kim, S.C.; Jiang, C.; Barma, D.; Kwon, S.W.; Cheng, J.; Jaunbergs, J.; Weinbaum, C.; Tamanoi, F.; Falck, J.; et al. A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12479–12484. [Google Scholar] [CrossRef]
- Sprung, R.; Nandi, A.; Chen, Y.; Kim, S.C.; Barma, D.; Falck, J.R.; Zhao, Y. Tagging-via-substrate strategy for probing O-GlcNAc modified proteins. J. Proteome Res. 2005, 4, 950–957. [Google Scholar] [CrossRef]
- Kostiuk, M.A.; Corvi, M.M.; Keller, B.O.; Plummer, G.; Prescher, J.A.; Hangauer, M.J.; Bertozzi, C.R.; Rajaiah, G.; Falck, J.R.; Berthiaume, L.G. Identification of palmitoylated mitochondrial proteins using a bio-orthogonal azido-palmitate analogue. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 22, 721–732. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.D.O.; Vilas, G.L.; Prescher, J.A.; Rajaiah, G.; Falck, J.R.; Bertozzi, C.R.; Berthiaume, L.G. Rapid detection, discovery, and identification of post-translationally myristoylated proteins during apoptosis using a bio-orthogonal azidomyristate analog. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 22, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Tsiatsiani, L.; Heck, A.J. Proteomics beyond trypsin. FEBS J. 2015, 282, 2612–2626. [Google Scholar] [CrossRef]
- Guo, X.; Trudgian, D.C.; Lemoff, A.; Yadavalli, S.; Mirzaei, H. Confetti: A multiprotease map of the HeLa proteome for comprehensive proteomics. Mol. Cell. Proteom. 2014, 13, 1573–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.G.; Kim, S.; Maltby, D.A.; Ghassemian, M.; Bandeira, N.; Komives, E.A. Expanding proteome coverage with orthogonal-specificity α-lytic proteases. Mol. Cell. Proteom. 2014, 13, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Bian, Y.; Ye, M.; Song, C.; Cheng, K.; Wang, C.; Wei, X.; Zhu, J.; Chen, R.; Wang, F.; Zou, H. Improve the coverage for the analysis of phosphoproteome of HeLa cells by a tandem digestion approach. J. Proteome Res. 2012, 11, 2828–2837. [Google Scholar] [CrossRef] [PubMed]
- Huesgen, P.F.; Lange, P.F.; Rogers, L.D.; Solis, N.; Eckhard, U.; Kleifeld, O.; Goulas, T.; Gomis-Ruth, F.X.; Overall, C.M. LysargiNase mirrors trypsin for protein C-terminal and methylation-site identification. Nat. Methods 2014, 12, 55–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J., 3rd. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2002, 346, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Kyle, R.A.; Buadi, F.K. Advances in the diagnosis, classification, risk stratification, and management of monoclonal gammopathy of undetermined significance: Implications for recategorizing disease entities in the presence of evolving scientific evidence. Mayo Clin. Proc. 2010, 85, 945–948. [Google Scholar] [CrossRef] [Green Version]
- Murray, D.L.; Seningen, J.L.; Dispenzieri, A.; Snyder, M.R.; Kyle, R.A.; Rajkumar, S.V.; Katzmann, J.A. Laboratory Persistence and Clinical Progression of Small Monoclonal Abnormalities. Am. J. Clin. Pathol. 2012, 138, 609–613. [Google Scholar] [CrossRef] [Green Version]
- An, G.; Acharya, C.; Feng, X.; Wen, K.; Zhong, M.; Zhang, L.; Munshi, N.C.; Qiu, L.; Tai, Y.-T.; Anderson, K.C. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: Therapeutic implication. Blood 2016, 128, 1590–1603. [Google Scholar] [CrossRef] [Green Version]
- Seckinger, A.; Meiβner, T.E.; Moreaux, J.; Depeweg, D.; Hillengass, J.; Hose, K.; Rème, T.; Rösen-Wolff, A.; Jauch, A.; Schnettler, R.; et al. Clinical and prognostic role of annexin A2 in multiple myeloma. Blood 2012, 120, 1087–1094. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Boswell, E.N.; Glavey, S.; Sacco, A.; Maiso, P.; Mishima, Y.; Aljaway, Y.; Banwait, R.; Zhang, W.; Zhang, Y.; et al. Mirna Expression Profiling and Proteomic Analysis Of Circulating Exosomes From Multiple Myeloma Patients. Blood 2013, 122, 3086. [Google Scholar] [CrossRef]
- Mailankody, S.; Devlin, S.M.; Korde, N.; Lendvai, N.; Lesokhin, A.; Landau, H.; Hassoun, H.; Ballagi, A.; Ekman, D.; Chung, D.J.; et al. Proteomic profiling in plasma cell disorders: A feasibility study. Leuk. Lymphoma 2016, 58, 1757–1759. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Witzig, T.E.; Timm, M.; Haug, J.; Wellik, L.; Fonseca, R.; Greipp, P.R.; Rajkumar, S.V. Expression of VEGF and its receptors by myeloma cells. Leukemia 2003, 17, 2025–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Brioli, A.; Johnson, D.C.; Kaiser, M.F.; Mirabella, F.; Lopez-Corral, L.; Humphray, S.; Murray, L.; et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia 2013, 28, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispenzieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N. Engl. J. Med. 2007, 356, 2582–2590. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Morgan, G.J. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer 2017, 17, 543–556. [Google Scholar] [CrossRef]
- Lussier, T.; Schoebe, N.; Mai, S. Risk Stratification and Treatment in Smoldering Multiple Myeloma. Cells 2021, 11, 130. [Google Scholar] [CrossRef]
- Fernandez, N.; Perumal, D.; Rahman, A.; Kim-Schulze, S.; Yesil, J.; Auclair, D.; Adams, H., 3rd; Parekh, S.; Gnjatic, S.; Cho, H.J. High Dimensional Immune Profiling of Smoldering Multiple Myeloma Distinguishes Distinct Tumor Microenvironments. Clin. Lymphoma Myeloma Leuk. 2022, 22, 853–862. [Google Scholar] [CrossRef]
- Chen, H.; Gordon, M.S.; Campbell, R.A.; Li, M.; Wang, C.S.; Lee, H.J.; Sanchez, E.; Manyak, S.J.; Gui, D.; Shalitin, D.; et al. Pleiotrophin is highly expressed by myeloma cells and promotes myeloma tumor growth. Blood 2007, 110, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Yeh, H.S.; Chen, H.; Manyak, S.J.; Swift, R.A.; Campbell, R.A.; Wang, C.; Li, M.; Lee, H.J.; Waterman, G.; Gordon, M.S.; et al. Serum pleiotrophin levels are elevated in multiple myeloma patients and correlate with disease status. Br. J. Haematol. 2006, 133, 526–529. [Google Scholar] [CrossRef]
- Lightbody, E.D.; Keshishian, H.; El-Khoury, H.; Dutta, A.K.; Barr, H.; Barbagallo, J.; Udeshi, N.; Agius, M.P.; Su, N.K.; Boehner, C.J.; et al. High-Throughput Plasma Proteomic Profiling to Identify Candidate High-Risk Disease Biomarkers in Precursor Multiple Myeloma Patients. Blood 2022, 140, 4303–4304. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, M.; He, P.; Chen, Y.; Wang, X.; Zhang, M. Identification of glutathione S-transferase π 1 as a prognostic proteomic biomarker for multiple myeloma using proteomic profiling. Oncol. Lett. 2020, 19, 2153–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.T.; Zhang, X.J.; Wang, H.; Yang, G.; Xu, X.F. IgD-λ multiple myeloma accompanying with elevated AFP level: A case report and literature review. Int. J. Clin. Exp. Med. 2018, 11, 5176–5180. [Google Scholar]
- Sahu, A.; Lambris, J.D. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol. Rev. 2001, 180, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, B.; Nilsson Ekdahl, K. The tick-over theory revisited: Is C3 a contact-activated protein? Immunobiology 2012, 217, 1106–1110. [Google Scholar] [CrossRef]
- Wlazlo, N.; van Greevenbroek, M.M.; Ferreira, I.; Jansen, E.J.; Feskens, E.J.; van der Kallen, C.J.; Schalkwijk, C.G.; Bravenboer, B.; Stehouwer, C.D. Low-grade inflammation and insulin resistance independently explain substantial parts of the association between body fat and serum C3: The CODAM study. Metabolism 2012, 61, 1787–1796. [Google Scholar] [CrossRef]
- Van Timmeren, M.M.; Chen, M.; Heeringa, P. Review article: Pathogenic role of complement activation in anti-neutrophil cytoplasmic auto-antibody-associated vasculitis. Nephrology 2009, 14, 16–25. [Google Scholar] [CrossRef]
- Xia, S.; Wang, C.; Postma, E.L.; Yang, Y.; Ni, X.; Zhan, W. Fibronectin 1 promotes migration and invasion of papillary thyroid cancer and predicts papillary thyroid cancer lymph node metastasis. OncoTargets Ther. 2017, 10, 1743–1755. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.J.; Lee, S.W.; Lin, L.C.; Chen, T.J.; Chang, I.W.; Hsu, H.P.; Chang, K.Y.; Huang, H.Y.; Li, C.F. Fibronectin overexpression is associated with latent membrane protein 1 expression and has independent prognostic value for nasopharyngeal carcinoma. Tumor Biol. J. Int. Soc. Oncodev. Biol. Med. 2013, 35, 1703–1712. [Google Scholar] [CrossRef]
- Sponziello, M.; Rosignolo, F.; Celano, M.; Maggisano, V.; Pecce, V.; De Rose, R.F.; Lombardo, G.E.; Durante, C.; Filetti, S.; Damante, G.; et al. Fibronectin-1 expression is increased in aggressive thyroid cancer and favors the migration and invasion of cancer cells. Mol. Cell. Endocrinol. 2016, 431, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Liu, Y.; Qin, R.; Liu, D.; Feng, Q. Silence of fibronectin 1 increases cisplatin sensitivity of non-small cell lung cancer cell line. Biochem. Biophys. Res. Commun. 2016, 476, 35–41. [Google Scholar] [CrossRef]
- Yi, W.; Xiao, E.; Ding, R.; Luo, P.; Yang, Y. High expression of fibronectin is associated with poor prognosis, cell proliferation and malignancy via the NF-κB/p53-apoptosis signaling pathway in colorectal cancer. Oncol. Rep. 2016, 36, 3145–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerhammar, F.; Ceder, R.; Garvin, S.; Grénman, R.; Grafström, R.C.; Roberg, K. Fibronectin 1 is a potential biomarker for radioresistance in head and neck squamous cell carcinoma. Cancer Biol. Ther. 2010, 10, 1244–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tancred, T.M.; Belch, A.R.; Reiman, T.; Pilarski, L.M.; Kirshner, J. Altered expression of fibronectin and collagens I and IV in multiple myeloma and monoclonal gammopathy of undetermined significance. J. Histochem. Cytochem. 2008, 57, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [Green Version]
- Strange, R.C.; Spiteri, M.A.; Ramachandran, S.; Fryer, A.A. Glutathione-S-transferase family of enzymes. Mutat. Res. 2001, 482, 21–26. [Google Scholar] [CrossRef]
- Adler, V.; Yin, Z.; Fuchs, S.Y.; Benezra, M.; Rosario, L.; Tew, K.D.; Pincus, M.R.; Sardana, M.; Henderson, C.J.; Wolf, C.R.; et al. Regulation of JNK signaling by GSTp. EMBO J. 1999, 18, 1321–1334. [Google Scholar] [CrossRef]
- Ding, H.; Liu, W.; Yu, X.; Wang, L.; Shao, L.; Yi, W. Risk association of meningiomas with MTHFR C677T and GSTs polymorphisms: A meta-analysis. Int. J. Clin. Exp. Med. 2014, 7, 3904–3914. [Google Scholar]
- Tew, K.D.; Manevich, Y.; Grek, C.; Xiong, Y.; Uys, J.; Townsend, D.M. The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free. Radic. Biol. Med. 2011, 51, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Emadi, A.; Karp, J.E.; Goodman, A.; Ball, E.D.; Megías-Vericat, J.E.; Montesinos, P.; Herrero, M.J.; Bosó, V.; Martínez-Cuadrón, D.; Poveda, J.L.; et al. The clinically relevant pharmacogenomic changes in acute myelogenous leukemia. Pharmacogenomics 2012, 13, 1257–1269. [Google Scholar] [CrossRef] [Green Version]
- Fernando, R.C.; de Carvalho, F.; Leme, A.F.P.; Colleoni, G.W.B. Tumor Microenvironment Proteomics: Lessons From Multiple Myeloma. Front. Oncol. 2021, 11, 563384. [Google Scholar] [CrossRef]
- Dytfeld, D.; Rosebeck, S.; Kandarpa, M.; Mayampurath, A.; Mellacheruvu, D.; Alonge, M.M.; Ngoka, L.; Jasielec, J.; Richardson, P.G.; Volchenboum, S.; et al. Proteomic profiling of naïve multiple myeloma patient plasma cells identifies pathways associated with favourable response to bortezomib-based treatment regimens. Br. J. Haematol. 2015, 170, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Storti, P.; Donofrio, G.; Marchica, V.; Guasco, D.; Todoerti, K.; Airoldi, I.; Anderson, J.; Bolzoni, M.; Ferri, V.; Martella, E.; et al. Galectin-1 Is Highly Expressed By Myeloma Cells and the Bone Marrow Microenvironment and Its Suppression Delineates a New Therapeutic in Vitro and in Vivo Strategy in Multiple Myeloma. Blood 2014, 124, 3373. [Google Scholar] [CrossRef]
- Munshi, N.C.; Hideshima, T.; Carrasco, D.; Shammas, M.; Auclair, D.; Davies, F.; Mitsiades, N.; Mitsiades, C.; Kim, R.S.; Li, C.; et al. Identification of genes modulated in multiple myeloma using genetically identical twin samples. Blood 2004, 103, 1799–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Maeda, H.; Reddy, S.V.; Kurihara, N.; Leach, R.; Anderson, J.L.; Roodman, G.D. Cloning and Characterization of the Annexin II Receptor on Human Marrow Stromal Cells. J. Biol. Chem. 2006, 281, 30542–30550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, S.; Kurihara, N.; Shiozawa, Y.; Joseph, J.; Taichman, R.; Galson, D.L.; Roodman, G.D. Annexin II interactions with the annexin II receptor enhance multiple myeloma cell adhesion and growth in the bone marrow microenvironment. Blood 2012, 119, 1888–1896. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.C.; Sharma, M. The role of annexin II in angiogenesis and tumor progression: A potential therapeutic target. Curr. Pharm. Des. 2007, 13, 3568–3575. [Google Scholar] [CrossRef]
- Glavey, S.V.; Naba, A.; Manier, S.; Clauser, K.; Tahri, S.; Park, J.; Reagan, M.R.; Moschetta, M.; Mishima, Y.; Gambella, M.; et al. Proteomic characterization of human multiple myeloma bone marrow extracellular matrix. Leukemia 2017, 31, 2426–2434. [Google Scholar] [CrossRef]
- Menaa, C.; Devlin, R.D.; Reddy, S.V.; Gazitt, Y.; Choi, S.J.; Roodman, G.D. Annexin II increases osteoclast formation by stimulating the proliferation of osteoclast precursors in human marrow cultures. J. Clin. Investig. 1999, 103, 1605–1613. [Google Scholar] [CrossRef] [Green Version]
- Storti, P.; Marchica, V.; Airoldi, I.; Donofrio, G.; Fiorini, E.; Ferri, V.; Guasco, D.; Todoerti, K.; Silbermann, R.; Anderson, J.L.; et al. Galectin-1 suppression delineates a new strategy to inhibit myeloma-induced angiogenesis and tumoral growth in vivo. Leukemia 2016, 30, 2351–2363. [Google Scholar] [CrossRef]
- Andersen, M.N.; Ludvigsen, M.; Abildgaard, N.; Petruskevicius, I.; Hjortebjerg, R.; Bjerre, M.; Honoré, B.; Møller, H.J.; Andersen, N.F. Serum galectin-1 in patients with multiple myeloma: Associations with survival, angiogenesis, and biomarkers of macrophage activation. OncoTargets Ther. 2017, 10, 1977–1982. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Song, Y.; Du, T.; Chauhan, D.; Anderson, K.C. Preclinical validation of Alpha-Enolase (ENO1) as a novel immunometabolic target in multiple myeloma. Oncogene 2020, 39, 2786–2796. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.L.; Ulanet, D.B.; Pirman, D.; Mahoney, C.E.; Coco, J.; Si, Y.; Chen, Y.; Huang, L.; Ren, J.; Choe, S.; et al. Differential Aspartate Usage Identifies a Subset of Cancer Cells Particularly Dependent on OGDH. Cell Rep. 2016, 17, 876–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Feng, Y.; Li, F.; Jia, Y.; Peng, Y.; Zhao, W.; Hu, J.; He, A. lncRNA ST3GAL6-AS1 promotes invasion by inhibiting hnRNPA2B1-mediated ST3GAL6 expression in multiple myeloma. Int. J. Oncol. 2021, 58, 1. [Google Scholar] [CrossRef]
- Chanukuppa, V.; Taware, R.; Taunk, K.; Chatterjee, T.; Sharma, S.; Somasundaram, V.; Rashid, F.; Malakar, D.; Santra, M.K.; Rapole, S. Proteomic Alterations in Multiple Myeloma: A Comprehensive Study Using Bone Marrow Interstitial Fluid and Serum Samples. Front. Oncol. 2021, 10, 566804. [Google Scholar] [CrossRef] [PubMed]
- Chanukuppa, V.; Paul, D.; Taunk, K.; Chatterjee, T.; Sharma, S.; Shirolkar, A.; Islam, S.; Santra, M.K.; Rapole, S. Proteomics and functional study reveal marginal zone B and B1 cell specific protein as a candidate marker of multiple myeloma. Int. J. Oncol. 2020, 57, 325–337. [Google Scholar] [CrossRef]
- Apipongrat, D.; Roytrakul, S.; Prayongratana, K.; Charoenpitakchai, M.; Intharanut, K.; Laoruangroj, C.; Silpsamrit, P.; Nathalang, O. Serum proteomic profiling reveals MTA2 and AGO2 as potential prognostic biomarkers associated with disease activity and adverse outcomes in multiple myeloma. PLoS ONE 2022, 17, e0278464. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, A.; Jakubowiak, A.; Hari, M.; Nesvizhskii, A. Proteomic Alterations Caused by Treatment with Velcade, Doxorubicin, and Dexamethasone in Myeloma. Clin. Lymphoma Myeloma Leuk. 2009, 9, S139. [Google Scholar] [CrossRef]
- Walker, Z.J.; Idler, B.M.; Davis, L.N.; Stevens, B.M.; VanWyngarden, M.J.; Ohlstrom, D.; Bearrows, S.C.; Hammes, A.; Smith, C.A.; Jordan, C.T.; et al. Exploiting Protein Translation Dependence in Multiple Myeloma with Omacetaxine-Based Therapy. Clin. Cancer Res. 2021, 27, 819–830. [Google Scholar] [CrossRef]
- Köse, M.C.; Lejeune, M.; Gou, M.J.; Duray, E.; Cobraiville, G.; Foguenne, J.; Gothot, A.; Beguin, Y.; Fillet, M.; Caers, J. Integrative Analysis of Proteomics and Transcriptomics Reveals the Etrb As Novel Single Target and New Combinatorial Targets for Multiple Myeloma. Blood 2022, 140, 4219. [Google Scholar] [CrossRef]
- Janker, L.; Mayer, R.L.; Bileck, A.; Kreutz, D.; Mader, J.C.; Utpatel, K.; Heudobler, D.; Agis, H.; Gerner, C.; Slany, A. Metabolic, Anti-apoptotic and Immune Evasion Strategies of Primary Human Myeloma Cells Indicate Adaptations to Hypoxia. Mol. Cell. Proteom. 2019, 18, 936–953. [Google Scholar] [CrossRef]
- Xiao, C.L.; Zhang, Z.P.; Xiong, S.; Lu, C.H.; Wei, H.P.; Zeng, H.L.; Liu, Z.; Zhang, X.E.; Ge, F. Comparative proteomic analysis to discover potential therapeutic targets in human multiple myeloma. Proteom. Clin. Appl. 2009, 3, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Wittenmayer, N.; Jandrig, B.; Rothkegel, M.; Schlüter, K.; Arnold, W.; Haensch, W.; Scherneck, S.; Jockusch, B.M.; Mullins, R.D.; Kelleher, J.F.; et al. Tumor suppressor activity of profilin requires a functional actin binding site. Mol. Biol. Cell 2004, 15, 1600–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witke, W. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 2004, 14, 461–469. [Google Scholar] [CrossRef]
- Choi, Y.H. Proteasome-mediated degradation of BRCA1 protein in MCF-7 human breast cancer cells. Int. J. Oncol. 2001, 19, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Kumatori, A.; Tanaka, K.; Inamura, N.; Sone, S.; Ogura, T.; Matsumoto, T.; Tachikawa, T.; Shin, S.; Ichihara, A. Abnormally high expression of proteasomes in human leukemic cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7071–7075. [Google Scholar] [CrossRef]
- Kanayama, H.; Tanaka, K.; Aki, M.; Kagawa, S.; Miyaji, H.; Satoh, M.; Okada, F.; Sato, S.; Shimbara, N.; Ichihara, A. Changes in expressions of proteasome and ubiquitin genes in human renal cancer cells. Cancer Res 1991, 51, 6677–6685. [Google Scholar] [PubMed]
- Nishinaka, Y.; Nakamura, H.; Masutani, H.; Yodoi, J. Redox control of cellular function by thioredoxin; a new therapeutic direction in host defence. Arch. Immunol. Ther. Exp. 2001, 49, 285–292. [Google Scholar]
- Kuku, I.; Aydogdu, I.; Bayraktar, N.; Kaya, E.; Akyol, O.; Erkurt, M.A. Oxidant/antioxidant parameters and their relationship with medical treatment in multiple myeloma. Cell Biochem. Funct. 2005, 23, 47–50. [Google Scholar] [CrossRef]
- Rescher, U.; Gerke, V. Annexins–unique membrane binding proteins with diverse functions. J. Cell Sci. 2004, 117, 2631–2639. [Google Scholar] [CrossRef] [Green Version]
- Mansour, A.; Wakkach, A.; Blin-Wakkach, C. Emerging Roles of Osteoclasts in the Modulation of Bone Microenvironment and Immune Suppression in Multiple Myeloma. Front. Immunol. 2017, 8, 954. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Epstein, J.; Suva, L.J. Biomarkers that discriminate multiple myeloma patients with or without skeletal involvement detected using SELDI-TOF mass spectrometry and statistical and machine learning tools. Dis. Markers 2006, 22, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, P.; Hayes, C.; Ting, K.R.; Hameed, A.; Meiller, J.; Mitsiades, C.; Anderson, K.C.; Clynes, M.; Clarke, C.; Richardson, P.; et al. Identification of proteins found to be significantly altered when comparing the serum proteome from Multiple Myeloma patients with varying degrees of bone disease. BMC Genom. 2014, 15, 904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slany, A.; Haudek-Prinz, V.; Meshcheryakova, A.; Bileck, A.; Lamm, W.; Zielinski, C.; Gerner, C.; Drach, J. Extracellular matrix remodeling by bone marrow fibroblast-like cells correlates with disease progression in multiple myeloma. J. Proteome Res. 2013, 13, 844–854. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Chen, Y.; Quan, L.; Jia, C.; Guo, Y.; Wang, X.; Zhang, Y.; Jin, Y.; Liu, A. Proteomics-Based Approach Reveals the Involvement of SERPINB9 in Recurrent and Relapsed Multiple Myeloma. J. Proteome Res. 2021, 20, 2673–2686. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P.; de Jong, J.; Peltenburg, L.T.; Verdegaal, E.M.; Gorter, A.; Bres, S.A.; Franken, K.L.; Hahne, M.; Albar, J.P.; Melief, C.J.; et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 11515–11520. [Google Scholar] [CrossRef]
- Bird, C.H.; Sutton, V.R.; Sun, J.; Hirst, C.E.; Novak, A.; Kumar, S.; Trapani, J.A.; Bird, P.I. Selective regulation of apoptosis: The cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol. Cell. Biol. 1998, 18, 6387–6398. [Google Scholar] [CrossRef] [Green Version]
- Pinkoski, M.J.; Waterhouse, N.J.; Heibein, J.A.; Wolf, B.B.; Kuwana, T.; Goldstein, J.C.; Newmeyer, D.D.; Bleackley, R.C.; Green, D.R. Granzyme B-mediated apoptosis proceeds predominantly through a Bcl-2-inhibitable mitochondrial pathway. J. Biol. Chem. 2001, 276, 12060–12067. [Google Scholar] [CrossRef] [Green Version]
- Hirst, C.E.; Buzza, M.S.; Bird, C.H.; Warren, H.S.; Cameron, P.U.; Zhang, M.; Ashton-Rickardt, P.G.; Bird, P.I. The intracellular granzyme B inhibitor, proteinase inhibitor 9, is up-regulated during accessory cell maturation and effector cell degranulation, and its overexpression enhances CTL potency. J. Immunol. 2003, 170, 805–815. [Google Scholar] [CrossRef]
- Medema, J.P.; Schuurhuis, D.H.; Rea, D.; van Tongeren, J.; de Jong, J.; Bres, S.A.; Laban, S.; Toes, R.E.; Toebes, M.; Schumacher, T.N.; et al. Expression of the serpin serine protease inhibitor 6 protects dendritic cells from cytotoxic T lymphocyte–induced apoptosis: Differential modulation by T helper type 1 and type 2 cells. J. Exp. Med. 2001, 194, 657–668. [Google Scholar] [CrossRef]
- Kaiserman, D.; Bird, P.I. Control of granzymes by serpins. Cell Death Differ. 2009, 17, 586–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Łuczak, M.; Kubicki, T.; Rzetelska, Z.; Szczepaniak, T.; Przybyłowicz-Chalecka, A.; Ratajczak, B.; Czerwińska-Rybak, J.; Nowicki, A.; Joks, M.; Jakubowiak, A.; et al. Comparative proteomic profiling of sera from patients with refractory multiple myeloma reveals potential biomarkers predicting response to bortezomib-based therapy. Pol. Arch. Intern. Med. 2017, 127, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, X.; He, D.; Zheng, G.; Yang, Y.; Han, X.; Li, Y.; Zhao, Y.; Wu, W.; Chen, Q.; Zhang, E.; et al. Analysis of High-Risk Extramedullary Relapse Factors in Newly Diagnosed MM Patients. Cancers 2022, 14, 6106. [Google Scholar] [CrossRef]
- Bladé, J.; Beksac, M.; Caers, J.; Jurczyszyn, A.; von Lilienfeld-Toal, M.; Moreau, P.; Rasche, L.; Rosiñol, L.; Usmani, S.Z.; Zamagni, E.; et al. Extramedullary disease in multiple myeloma: A systematic literature review. Blood Cancer J. 2022, 12, 45. [Google Scholar] [CrossRef]
- Zatula, A.; Dikic, A.; Mulder, C.; Sharma, A.; Vågbø, C.B.; Sousa, M.M.L.; Waage, A.; Slupphaug, G. Proteome alterations associated with transformation of multiple myeloma to secondary plasma cell leukemia. Oncotarget 2016, 8, 19427–19442. [Google Scholar] [CrossRef]
- Dunphy, K.; Bazou, D.; Dowling, P.; O’Gorman, P. Proteomic Characterisation of the Plasma Proteome in Extramedullary Multiple Myeloma Identifies Potential Prognostic Biomarkers. Blood 2022, 140, 10058–10059. [Google Scholar] [CrossRef]
- Dunphy, K.; Bazou, D.; Henry, M.; Meleady, P.; Dowling, P.; O’Gorman, P. Characterisation of the Tumour Proteome in Primary Extramedullary Multiple Myeloma Identifies Key Proteins Associated with Transendothelial Migration. Blood 2021, 138, 2665. [Google Scholar] [CrossRef]
- Wallington-Beddoe, C.T.; Sobieraj-Teague, M.; Kuss, B.J.; Pitson, S. Resistance to proteasome inhibitors and other targeted therapies in myeloma. Br. J. Haematol. 2018, 182, 11–28. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Chen, Y.; Saha, M.N.; Chen, J.; Evans, K.; Qiu, L.; Reece, D.; Chen, G.A.; Chang, H. Targeting phospho-MARCKS overcomes drug-resistance and induces antitumor activity in preclinical models of multiple myeloma. Leukemia 2014, 29, 715–726. [Google Scholar] [CrossRef]
- Ting, K.R.; Henry, M.; Meiller, J.; Larkin, A.; Clynes, M.; Meleady, P.; Bazou, D.; Dowling, P.; O’Gorman, P. Novel panel of protein biomarkers to predict response to bortezomib-containing induction regimens in multiple myeloma patients. BBA Clin. 2017, 8, 28–34. [Google Scholar] [CrossRef]
- Hsieh, F.Y.; Tengstrand, E.; Pekol, T.M.; Guerciolini, R.; Miwa, G. Elucidation of potential bortezomib response markers in mutliple myeloma patients. J. Pharm. Biomed. Anal. 2009, 49, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Dytfeld, D.; Luczak, M.; Wrobel, T.; Usnarska-Zubkiewicz, L.; Brzezniakiewicz, K.; Jamroziak, K.; Giannopoulos, K.; Przybylowicz-Chalecka, A.; Ratajczak, B.; Czerwinska-Rybak, J.; et al. Comparative proteomic profiling of refractory/relapsed multiple myeloma reveals biomarkers involved in resistance to bortezomib-based therapy. Oncotarget 2016, 7, 56726–56736. [Google Scholar] [CrossRef] [Green Version]
- Frassanito, M.A.; De Veirman, K.; Desantis, V.; Di Marzo, L.; Vergara, D.; Ruggieri, S.; Annese, T.; Nico, B.; Menu, E.; Catacchio, I.; et al. Halting pro-survival autophagy by TGFβ inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia 2015, 30, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.R.; Wu, Z.; Chauhan, D.; Anderson, K.C.; Peng, J. A nano ultra-performance liquid chromatography–high resolution mass spectrometry approach for global metabolomic profiling and case study on drug-resistant multiple myeloma. Anal. Chem. 2014, 86, 3667–3675. [Google Scholar] [CrossRef] [PubMed]
- Maiso, P.; Huynh, D.; Moschetta, M.; Sacco, A.; Aljawai, Y.; Mishima, Y.; Asara, J.M.; Roccaro, A.M.; Kimmelman, A.C.; Ghobrial, I.M. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res 2015, 75, 2071–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, Y.L.D.; Ramberger, E.; Bohl, S.R.; Dolnik, A.; Steinebach, C.; Conrad, T.; Müller, S.; Popp, O.; Kull, M.; Haji, M.; et al. Proteomic profiling reveals CDK6 upregulation as a targetable resistance mechanism for lenalidomide in multiple myeloma. Nat. Commun. 2022, 13, 1009. [Google Scholar] [CrossRef] [PubMed]
- Chanukuppa, V.; Paul, D.; Taunk, K.; Chatterjee, T.; Sharma, S.; Kumar, S.; Santra, M.K.; Rapole, S. XPO1 is a critical player for bortezomib resistance in multiple myeloma: A quantitative proteomic approach. J. Proteom. 2019, 209, 103504. [Google Scholar] [CrossRef]
- Van der Watt, P.J.; Maske, C.P.; Hendricks, D.T.; Parker, M.I.; Denny, L.; Govender, D.; Birrer, M.J.; Leaner, V.D. The Karyopherin proteins, Crm1 and Karyopherin β1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int. J. Cancer 2009, 124, 1829–1840. [Google Scholar] [CrossRef]
- Stommel, J.M.; Marchenko, N.D.; Jimenez, G.S.; Moll, U.M.; Hope, T.J.; Wahl, G.M. A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999, 18, 1660–1672. [Google Scholar] [CrossRef] [Green Version]
- Nikolaev, A.Y.; Li, M.; Puskas, N.; Qin, J.; Gu, W. Parc: A cytoplasmic anchor for p53. Cell 2003, 112, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noske, A.; Weichert, W.; Niesporek, S.; Röske, A.; Buckendahl, A.C.; Koch, I.; Sehouli, J.; Dietel, M.; Denkert, C. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer 2008, 112, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Senapedis, W.; McCauley, D.; Baloglu, E.; Shacham, S.; Festuccia, C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J. Hematol. Oncol. 2014, 7, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muqbil, I.; Kauffman, M.; Shacham, S.; Mohammad, R.; Azmi, A. Understanding XPO1 Target Networks Using Systems Biology and Mathematical Modeling. Curr. Pharm. Des. 2014, 20, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Baek, H.B.; Lombard, A.P.; Libertini, S.J.; Fernandez-Rubio, A.; Vinall, R.; Gandour-Edwards, R.; Nakagawa, R.; Vidallo, K.; Nishida, K.; Siddiqui, S.; et al. XPO1 inhibition by selinexor induces potent cytotoxicity against high grade bladder malignancies. Oncotarget 2018, 9, 34567–34581. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.S.; Bedard, P.L.; Kuruvilla, J.; Siu, L.L.; Razak, A.R. Promising SINEs for embargoing nuclear–cytoplasmic export as an anticancer strategy. Cancer Discov. 2014, 4, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Vermeer, H.; Hendriks-Stegeman, B.I.; Van Der Burg, B.; Van Buul-Offers, S.C.; Jansen, M. Glucocorticoid-induced increase in lymphocytic FKBP51 messenger ribonucleic acid expression: A potential marker for glucocorticoid sensitivity, potency, and bioavailability. J. Clin. Endocrinol. Metab. 2003, 88, 277–284. [Google Scholar] [CrossRef]
- Rees-Unwin, K.S.; Craven, R.A.; Davenport, E.; Hanrahan, S.; Totty, N.F.; Dring, A.M.; Banks, R.E.; J. Morgan, G.; Davies, F.E. Proteomic evaluation of pathways associated with dexamethasone-mediated apoptosis and resistance in multiple myeloma. Br. J. Haematol. 2007, 139, 559–567. [Google Scholar] [CrossRef]
- Besse, L.; Soriano, G.; Li, N.; Besse, A.; Kraus, M.; Bader, J.; Silzle, T.C.; Meeuwenoord, N.; den Dulk, H.; Overkleeft, H.S.; et al. Proteasome Inhibitor-Adapted Myeloma Cells That Lack Proteasome Gene Mutations Resist Highly Efficient Proteasome Inhibition and Show Proteomic Alterations That Suggest Complex Metabolic Changes. Blood 2015, 126, 1278. [Google Scholar] [CrossRef]
- Rajpal, R.; Dowling, P.; Meiller, J.; Clarke, C.; Murphy, W.G.; O’Connor, R.; Kell, M.; Mitsiades, C.; Richardson, P.; Anderson, K.C.; et al. A novel panel of protein biomarkers for predicting response to thalidomide-based therapy in newly diagnosed multiple myeloma patients. Proteomics 2011, 11, 1391–1402. [Google Scholar] [CrossRef]
- Zhou, N.; Gutierrez-Uzquiza, A.; Zheng, X.Y.; Chang, R.; Vogl, D.T.; Garfall, A.L.; Bernabei, L.; Saraf, A.; Florens, L.; Washburn, M.P.; et al. RUNX proteins desensitize multiple myeloma to lenalidomide via protecting IKZFs from degradation. Leukemia 2019, 33, 2006–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harshman, S.W.; Canella, A.; Ciarlariello, P.D.; Agarwal, K.; Branson, O.E.; Rocci, A.; Cordero, H.; Phelps, M.A.; Hade, E.M.; Dubovsky, J.A.; et al. Proteomic characterization of circulating extracellular vesicles identifies novel serum myeloma associated markers. J. Proteom. 2016, 136, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertweck, M.K.; Erdfelder, F.; Kreuzer, K.-A. CD44 in hematological neoplasias. Ann. Hematol. 2011, 90, 493–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohwada, C.; Nakaseko, C.; Koizumi, M.; Takeuchi, M.; Ozawa, S.; Naito, M.; Tanaka, H.; Oda, K.; Cho, R.; Nishimura, M.; et al. CD44 and hyaluronan engagement promotes dexamethasone resistance in human myeloma cells. Eur. J. Haematol. 2008, 80, 245–250. [Google Scholar] [CrossRef]
- Harshman, S.W.; Canella, A.; Ciarlariello, P.D.; Rocci, A.; Agarwal, K.; Smith, E.M.; Talabere, T.; Efebera, Y.A.; Hofmeister, C.C.; Benson, D.M., Jr.; et al. Characterization of multiple myeloma vesicles by label-free relative quantitation. Proteomics 2013, 13, 3013–3029. [Google Scholar] [CrossRef]
- Ho, M.; Bianchi, G.; Anderson, K.C. Proteomics-inspired precision medicine for treating and understanding multiple myeloma. Expert Rev. Precis. Med. Drug Dev. 2020, 5, 67–85. [Google Scholar] [CrossRef]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade®: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Hambley, B.; Caimi, P.F.; William, B.M. Bortezomib for the treatment of mantle cell lymphoma: An update. Ther. Adv. Hematol. 2016, 7, 196–208. [Google Scholar] [CrossRef]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nature 2005, 7, 766–772. [Google Scholar] [CrossRef]
- Voorhees, P.M.; Orlowski, R.Z. The proteasome and proteasome inhibitors in cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 189–213. [Google Scholar] [CrossRef]
- Loke, C.; Mollee, P.; McPherson, I.; Walpole, E.; Yue, M.; Mutsando, H.; Wong, P.; Weston, H.; Tomlinson, R.; Hollingworth, S. Bortezomib use and outcomes for the treatment of multiple myeloma. Intern. Med. J. 2020, 50, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor Activity of PR-171, a Novel Irreversible Inhibitor of the Proteasome. Cancer Res 2007, 67, 6383–6391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.B.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groll, M.; Kim, K.B.; Kairies, N.; Huber, R.; Crews, C.M. Crystal Structure of Epoxomicin:20S Proteasome Reveals a Molecular Basis for Selectivity of α‘,β‘-Epoxyketone Proteasome Inhibitors. J. Am. Chem. Soc. 2000, 122, 1237–1238. [Google Scholar] [CrossRef]
- Badros, A.Z.; Vij, R.; Martin, T.; Zonder, J.A.; Kunkel, L.; Wang, Z.; Lee, S.; Wong, A.F.; Niesvizky, R. Carfilzomib in multiple myeloma patients with renal impairment: Pharmacokinetics and safety. Leukemia 2013, 27, 1707–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herndon, T.M.; Deisseroth, A.; Kaminskas, E.; Kane, R.C.; Koti, K.M.; Rothmann, M.D.; Habtemariam, B.; Bullock, J.; Bray, J.D.; Hawes, J.; et al. U.S. Food and drug administration approval: Carfilzomib for the treatment of multiple myeloma. Clin. Cancer Res. 2013, 19, 4559–4563. [Google Scholar] [CrossRef] [Green Version]
- Crawford, L.J.; Irvine, A.E. Targeting the ubiquitin proteasome system in haematological malignancies. Blood Rev. 2013, 27, 297–304. [Google Scholar] [CrossRef]
- Siegel, D.S.; Dimopoulos, M.A.; Ludwig, H.; Facon, T.; Goldschmidt, H.; Jakubowiak, A.; San-Miguel, J.; Obreja, M.; Blaedel, J.; Stewart, A.K. Improvement in Overall Survival With Carfilzomib, Lenalidomide, and Dexamethasone in Patients With Relapsed or Refractory Multiple Myeloma. J. Clin. Oncol. 2018, 36, 728–734. [Google Scholar] [CrossRef]
- Raedler, L.A. Kyprolis (Carfilzomib) Received New Indications as Combination Therapy for Use in Relapsed and/or Refractory Multiple Myeloma. Am. Health Drug Benefits 2016, 9, 93–96. [Google Scholar]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hájek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef]
- Brayer, J.; Baz, R. The potential of ixazomib, a second-generation proteasome inhibitor, in the treatment of multiple myeloma. Ther. Adv. Hematol. 2017, 8, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Minarik, J.; Pika, T.; Radocha, J.; Jungova, A.; Straub, J.; Jelinek, T.; Pour, L.; Pavlicek, P.; Mistrik, M.; Brozova, L.; et al. Survival benefit of ixazomib, lenalidomide and dexamethasone (IRD) over lenalidomide and dexamethasone (Rd) in relapsed and refractory multiple myeloma patients in routine clinical practice. BMC Cancer 2021, 21, 73. [Google Scholar] [CrossRef]
- Saltarella, I.; Desantis, V.; Melaccio, A.; Solimando, A.G.; Lamanuzzi, A.; Ria, R.; Storlazzi, C.T.; Mariggiò, M.A.; Vacca, A.; Frassanito, M.A. Mechanisms of Resistance to Anti-CD38 Daratumumab in Multiple Myeloma. Cells 2020, 9, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touzeau, C.; Moreau, P. Daratumumab for the treatment of multiple myeloma. Expert Opin. Biol. Ther. 2017, 17, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Zanwar, S.; Nandakumar, B.; Kumar, S. Immune-based therapies in the management of multiple myeloma. Blood Cancer J. 2020, 10, 84. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Sonneveld, P.; Hungria, V.; Nooka, A.K.; Estell, J.A.; Barreto, W.; Corradini, P.; Min, C.-K.; Medvedova, E.; Weisel, K.; et al. Daratumumab, Bortezomib, and Dexamethasone Versus Bortezomib and Dexamethasone in Patients With Previously Treated Multiple Myeloma: Three-year Follow-up of CASTOR. Clin. Lymphoma Myeloma Leuk. 2019, 20, 509–518. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Facon, T.; Kumar, S.K.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab, lenalidomide, and dexamethasone versus lenalidomide and dexamethasone alone in newly diagnosed multiple myeloma (MAIA): Overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 1582–1596. [Google Scholar] [CrossRef]
- Magen, H.; Muchtar, E. Elotuzumab: The first approved monoclonal antibody for multiple myeloma treatment. Ther. Adv. Hematol. 2016, 7, 187–195. [Google Scholar] [CrossRef]
- Collins, S.M.; Bakan, C.E.; Swartzel, G.D.; Hofmeister, C.C.; Efebera, Y.A.; Kwon, H.; Starling, G.C.; Ciarlariello, D.; Bhaskar, S.S.; Briercheck, E.L.; et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: Evidence for augmented NK cell function complementing ADCC. Cancer Immunol. Immunother. 2013, 62, 1841–1849. [Google Scholar] [CrossRef] [Green Version]
- Parrondo, R.D.; Paulus, A.; Ailawadhi, S. Updates in the Use of BCL-2-Family Small Molecule Inhibitors for the Treatment of Relapsed/Refractory Multiple Myeloma. Cancers 2022, 14, 3330. [Google Scholar] [CrossRef] [PubMed]
- Ehsan, H.; Wahab, A.; Shah, Z.; Sana, M.K.; Masood, A.; Rafae, A.; Hashmi, H. Role of Venetoclax in the Treatment of Relapsed and Refractory Multiple Myeloma. J. Hematol. 2021, 10, 89–97. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Method Description |
|---|---|
| 2-dimensional polyacrylamide gel electrophoresis (2D-PAGE) | The protein content of a sample is resolved on a gel in two dimensions according to mass and charge; the gels are stained, and the spot intensities in the samples are analyzed among the multiple gels. |
| 2D-DIGE | Each protein sample of interest is labelled with a different fluorophore (Cy3, Cy5, or Cy2) that binds covalently to the epsilon amino group of lysine residues. |
| Protein microarrays | Direct labelling or labelled secondary antibodies are used to identify bound proteins once targeted proteins in one sample bind to probes on a “forward” microarray, and vice versa for “reverse” microarrays. |
| Surface-enhanced laser desorption/ ionization time-of-flight mass spectrometry (SELDI-TOF MS) | Retention chromatography and mass spectrometry principles are combined, offering a fast, high-throughput, and relatively sensitive screening approach for complicated protein samples. Proteins can also be separated, detected, and analyzed at the femtomole level straight from biological materials. This allows for the discovery of many analytes and the analysis of many diverse samples while studying multiple biological variables at the same time. |
| Matrix-assisted laser desorption/ ionization-time of flight mass spectrometry (MALDI-TOF MS) | Application of a protein mixture onto a gold plate, desorption of proteins from the plate using laser energy, and determination of the protein masses, with comparison of peak intensities between several different samples. |
| Liquid Chromatography with tandem mass spectrometry (LC-MS-MS) | Separation of a mixture of peptides (derived from trypsin-catalyzed protein digestion) through one-, two-, or three-dimensional LC and determination of peptide masses through MS-MS. |
| Isotope-coded affinity tag (ICAT) | Chemical tagging of proteins on cysteine residues with a heavy or light stable isotope; after labelling samples are combined, proteins are digested with trypsin, and tagged peptides are extracted via affinity chromatography; both samples are then concomitantly analyzed using LC-MS-MS. |
| Multiplexed Isobaric Tagging Technology for Relative Quantitation (iTRAQ) | A shotgun-based quantitation technique. Employs a multiplexed isobaric chemical tagging reagent that enables the multiplexing of two to eight protein samples and generates identical MS-MS sequencing ions for all eight variants of the same derivatized tryptic peptide. |
| SILAC (Stable Isotope Labeling by Amino Acids in Cell Culture) | Encompasses the practice of cultivating cells in a medium that comprises isotopically labelled amino acids. The isotopes utilized are generally heavy variants of essential amino acids, such as 13C- or 15N-labelled versions. The process of labelling induces a mass shift in the proteome, thereby facilitating precise quantification and comparison of protein expression levels across diverse conditions or samples. |
| Immunohistochemistry (IHC) | A diagnostic technique for visualizing and detecting particular proteins within tissue specimens. This technique facilitates the analysis of protein localization, interaction, and expression patterns with a high degree of spatial resolution. |
| Flow cytometry | The process of scrutinizing and measuring diverse physical and chemical attributes of individual cells or particles in a fluid suspension; is commonly referred to as a cellular or particle analysis technique. The technique facilitates the concurrent assessment of various parameters such as cell surface markers, intracellular proteins, DNA content, and cell viability. |
| Aptamer-based proteomics platforms | A novel technological approach involving the utilization of aptamers, which are short, single-stranded nucleic acids, as affinity reagents for identifying and analyzing proteins. The process of systematic evolution of ligands by exponential enrichment (SELEX) is utilized to generate aptamers that exhibit high affinity and selectivity towards target proteins. These aptamers are designed to specifically bind to the target proteins. |
| Singleplex ELISA (enzyme-linked immunosorbent assay (ELISA) | Also referred to as standard or traditional ELISA; a common methodology employed for the identification and measurement of an exclusive protein within a specimen. |
| Multiplex ELISA | A novel revision of the conventional enzyme-linked immunosorbent assay (ELISA) that enables the concurrent identification and measurement of numerous proteins within a solitary specimen. The utilization of multiplex ELISA allows for the investigation of multiple analytes present in intricate biological specimens, thereby facilitating a more exhaustive evaluation in contrast to singleplex ELISA. |
| Cluster Id | Gene Count | Protein Names | |
|---|---|---|---|
 | Cluster 1 | 19 | ACBD3, ATP5L, COX14, COX6C, ECHS1, EDNRB, ENO1, GTF2F2, HIF1AN, ICAM2, LPCAT3, OGDH, PGK1, PRDX4, SLCO5A1, SLCO6A1, TAOK2, TPI1, TRAF2 |
 | Cluster 2 | 37 | AFP, AGO2, ANXA1, ANXA11, ANXA2, ANXA8, BAX, C10orf54, CASP10, CD46, FCRL5, FN1, GPX1, GSTP1, HSPA8, IL6R, IRF4, LGALS1, MIF, MTA1, MTA2, MYC, MZB1, PFN1, S100A11, S100A12, S100A9, S100B, SDC1, SDF4, SIGIRR, STAB1, STMN1, TNFRSF17, TP53, VEPH1, VHL |
 | Cluster 3 | 17 | C19orf10, CANX, DAPP1, IPO7, ITPR1, MT-CO2, PRAF2, PSMA1, PSMA5, PSME2, PTPN1, RAB7A, RCN1, SDF2L1, TP53I11, TXNDC11, TXNDC5 |
| Cluster | GO-Term | Biological Process | False Discovery Rate |
|---|---|---|---|
| 1 | GO:0030195 | Negative regulation of blood coagulation | 0.0020 |
| GO:0051241 | Negative regulation of multicellular organismal process | 0.0020 | |
| GO:0042730 | Fibrinolysis | 0.0129 | |
| GO:0034110 | Regulation of homotypic cell–cell adhesion | 0.0239 | |
| 2 | GO:0046034 | ATP metabolic process | 0.00044 |
| GO:0006096 | Oxidation–reduction process | 0.00054 | |
| GO:0055114 | Glycolytic process | 0.0015 | |
| GO:0006091 | Generation of precursor metabolites and energy | 0.0018 | |
| 3 | GO:0002474 | Antigen processing and presentation of peptide antigen via MHC Class I | 0.0044 |
| GO:0002478 | Antigen processing and presentation of exogenous peptide antigen | 0.0044 | |
| GO:0034976 | Response to endoplasmic reticulum stress | 0.0047 | |
| GO:0035967 | Cellular response to topologically incorrect protein | 0.0141 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismail, N.H.; Mussa, A.; Al-Khreisat, M.J.; Mohamed Yusoff, S.; Husin, A.; Johan, M.F. Proteomic Alteration in the Progression of Multiple Myeloma: A Comprehensive Review. Diagnostics 2023, 13, 2328. https://doi.org/10.3390/diagnostics13142328
Ismail NH, Mussa A, Al-Khreisat MJ, Mohamed Yusoff S, Husin A, Johan MF. Proteomic Alteration in the Progression of Multiple Myeloma: A Comprehensive Review. Diagnostics. 2023; 13(14):2328. https://doi.org/10.3390/diagnostics13142328
Chicago/Turabian StyleIsmail, Nor Hayati, Ali Mussa, Mutaz Jamal Al-Khreisat, Shafini Mohamed Yusoff, Azlan Husin, and Muhammad Farid Johan. 2023. "Proteomic Alteration in the Progression of Multiple Myeloma: A Comprehensive Review" Diagnostics 13, no. 14: 2328. https://doi.org/10.3390/diagnostics13142328