
The Role of VHL in the Development of von Hippel-Lindau Disease and Erythrocytosis


1
Medical Centre for Molecular Biology, Institute of Biochemistry and Molecular Genetics, Faculty of Medicine, University of Ljubljana, Vrazov trg 2, 1000 Ljubljana, Slovenia
2
Eye Hospital, University Medical Centre Ljubljana, Grabloviceva ulica 46, 1000 Ljubljana, Slovenia
*
Author to whom correspondence should be addressed.
Genes 2022, 13(2), 362; https://doi.org/10.3390/genes13020362
Submission received: 29 January 2022
/
Revised: 14 February 2022
/
Accepted: 15 February 2022
/
Published: 17 February 2022
(This article belongs to the Special Issue Genetics and Genomics of Erythrocytosis)
Abstract
:Von Hippel-Lindau disease (VHL disease or VHL syndrome) is a familial multisystem neoplastic syndrome stemming from germline disease-associated variants of the VHL tumor suppressor gene on chromosome 3. VHL is involved, through the EPO-VHL-HIF signaling axis, in oxygen sensing and adaptive response to hypoxia, as well as in numerous HIF-independent pathways. The diverse roles of VHL confirm its implication in several crucial cellular processes. VHL variations have been associated with the development of VHL disease and erythrocytosis. The association between genotypes and phenotypes still remains ambiguous for the majority of mutations. It appears that there is a distinction between erythrocytosis-causing VHL variations and VHL variations causing VHL disease with tumor development. Understanding the pathogenic effects of VHL variants might better predict the prognosis and optimize management of the patient.
1. Introduction
Von Hippel-Lindau disease (VHL disease or VHL syndrome) is a familial multisystem syndrome stemming from germline disease-associated variants of the VHL tumor suppressor gene on chromosome 3 [1]. It is an autosomal dominant neoplastic disorder in which multiple benign and malignant tumors, as well as cysts, develop in the central nervous system (the brain, spinal cord, retina, and inner ear) and visceral organs (kidney, adrenal gland, pancreas, epididymis, and broad ligament) [2,3]. Despite its classification as a dominant disorder, the most common pattern in hereditary VHL disease is the inheritance of a germline genetic variant (herein, mutation) in one allele, followed by a second somatic change, leading to loss of the second allele [2,4,5]. In approximately 20% of cases, VHL syndrome is sporadic, caused by a de novo genetic change that arises during the formation of reproductive cells, or very early during the embryogenesis [4,6,7]. After the identification of the VHL gene in 1993, the phenotype of VHL pathogenic genetic variations was expanded to include VHL disease, dominantly inherited familial pheochromocytoma, and autosomal recessive familial polycythemia [8,9,10]. Chuvash polycythemia, a secondary congenital erythrocytosis, was the first congenital erythrocytosis linked to a VHL mutation [8]. Since then, several other VHL mutations associated with erythrocytosis have been identified [11].
The aim of this review is to provide an overview of the role of VHL gene mutations leading to the development of VHL disease and erythrocytosis, and to present a case of a patient with VHL disease due to a de novo mutation, to illustrate the role of genetic testing in the management of patients with genetic disease.
2. VHL Canonical and Non-Canonical Functions
The von Hippel-Lindau tumor suppressor gene (VHL) is located on chromosome 3. Initially it was thought to be composed of three exons that encode the VHL protein [12,13]. Subsequent research revealed more complex transcription patterns, and an additional cryptic exon, resulting in several different functional VHL isoforms (Figure 1) [14,15]. Soon after its discovery, it was confirmed that the two most commonly isolated isoforms, VHL30 or VHL213 and VHL19 or VHL160, are both biologically active and are expressed in all tissues [16,17,18].
VHL30 is the longest isoform, with a molecular mass of 30 kDa. It has 213 amino acids and consists of three exons, whereas VHL19 has 160 amino acids and a molecular mass of 19 kDa [16]. Iliopoulos et al. demonstrated that VHL19 is generated from an internal translational start at methionine 54 [16]. Another protein product of the VHL gene, VHL172, which arises from alternative splicing that joins exons one and three while excluding exon two, was also detected [17].
Recently, Lenglet et al. identified transcripts containing a cryptic exon, E1′, located in intron one of the VHL gene, spliced either with exon one or exons two and three [15]. They speculated that a theoretical protein consisting of 193 amino acids could exist, containing 114 amino acids encoded by exon one and 79 amino acids encoded by the cryptic exon, E1′. Their findings were linked to the aberrant retention of this cryptic exon in patients with VHL disease and erythrocytosis.
The reference sequence collection currently displays three mRNA and protein reference variants, which differ in length and exon structure. The three representative isoforms consist of 213, 172, and 193 amino acids residues, respectively [19]. UniProt database describes three isoforms, which are produced by alternative splicing and alternative initiation. Isoform one corresponds to RefSeq isoform one (VHL30 or VHL213) and isoform two corresponds to RefSeq isoform two (VHL172), whereas isoform three corresponds to VHL19 (160 amino acids) and is produced from alternative initiation at methionine 54 [20].
The longest protein isoform has two main protein binding domains, β and α domain, which are preceded by N-terminal acidic disordered domain [21,22]. The β domain is composed of approximately 100 amino acids. This domain is rich in β strands, which form a β-sheet structure, and interacts with hydroxylated HIFs, RNA polymerase II, protein kinase C isoforms, and other proteins [23,24]. The shorter α domain contains binding sites for Elongin-B/C (BC box), Cullin-2 (Cul-2 box), p53, and other proteins [23]. The β domain contains another binding interface, interface C, which is important for VHL localization and binds TBP1 and EEF1A [23].
On the subcellular level, the strongest VHL protein expression was observed in the cytosol, nucleus, mitochondrion and endoplasmic reticulum [25]. Interestingly, Illiopoulus et al. noted that VHL, which is composed of 213 amino acids and corresponds to VHL30, localizes predominantly to cytosol, or is membrane-associated [16]. It was found only in low levels in the nucleus, whereas the 160-amino acid VHL (VHL19) was found in both the nucleus and cytosol. In contrast to the cytosolic membrane-bound VHL30, VHL19 was not associated with cell membranes [16].
RNA sequencing of 27 different normal tissues, from 95 individuals, revealed that VHL protein is ubiquitously expressed in most tissues [26]. However, protein expression studies showed that, despite the relatively high abundance of mRNA in some tissues, the levels of VHL were low or absent, indicating that complex mechanisms govern the mRNA expression, protein synthesis, and/or protein stability of VHL (https://www.proteinatlas.org/ENSG00000134086-VHL/tissue, available from v21.0 proteinatlas.org, accessed on 10 January 2022) [26,27]. This is consistent with general observations in mammalian tissues, where a number of genes exhibited very low correlations between mRNA expression levels and protein staining (Pearson’s r ̴ 0.40) [26,28].
The most important canonical role of VHL, through the EPO-VHL-HIF signaling axis, is its involvement in oxygen sensing and adaptive response to hypoxia (Figure 2) [29,30,31,32,33]. VHL associates with Elongin-B (ELOB) and Elongin-C (ELOC), forming a VBC complex, and, together with Cullin-2 (CUL2) and E3 ubiquitin-protein ligase RBX1 (RBX, Ring-box1), constitutes a functional E3 ubiquitin ligase that specifically recognizes hydroxylated Hypoxia inducible factor (HIF) subunit α, targeting it for proteasome degradation by ubiquitination [34,35,36,37,38,39]. The VHL protein serves as a substrate (e.g., HIF1α) recruitment protein [40,41]. When oxygen levels are normal (normoxic conditions), the two proline residues on the N-terminal (NOOD) and C-terminal (COOD) regions of HIF1α, P402, and P564, respectively, are hydroxylated by the non-heme Fe2+ -oxygen and α-ketogluratate-dependent dioxygenase superfamily of prolyl hydroxylases (PHD1, PHD2, and PHD3) [42,43]. In vitro PHD2 catalyzes the hydroxylation of proline in COOD peptides more efficiently than the NOOD peptide proline, whereas PHD3 specifically hydroxylates only COOD proline [44].
The efficiencies of P402 and P564 hydroxylation differ, and when the level of oxygen falls, the hydroxylation of HIF1α prolines is gradually diminished, with P402 more affected than P564 [45,46]. VHL recognizes both hydroxylated prolines with similar binding affinities, resulting in interaction between HIF1α and VBC complex in a 1:2 stoichiometric ratio [47]. HIF1α can therefore bind two VBC complexes or one VBC complex, according to the status of P402 and P564 hydroxylation, at different oxygen levels [45,46,47]. The decrease in VHL targets could lead to gradual increase in HIF1α.
Additionally, in normoxia (normal levels of oxygen), the asparaginyl residue N803 is hydroxylated by factor inhibiting HIF (FIH) or asparaginyl hydroxylase, preventing HIF1α interaction with transcriptional coactivator p300 (EP300) [48,49]. Under low levels of oxygen (hypoxia), HIF1α-specific prolyl and asparaginyl residues are not hydroxylated by PHDs and FIH, respectively. Non-hydroxylated HIF1α subunits can enter the nucleus through specific importins, where they form heterodimeric complexes with Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT, also known as Hypoxia-inducible factor 1, β subunit, HIF1β) [43]. The HIF1α-ARNT complexes act as transcriptional regulators, and bind to consensus 5′-(A/G)CGTG-3′ sequence or HIF ancillary sequences 5′-CAGGT-3′ in hypoxia response elements (HRE), within promoters of several genes involved in adaptive response to hypoxia [42,43].
VHL is also involved in numerous HIF-independent pathways, including protein stabilization [50,51], regulation microtubule stability, and ciliogenesis, as well as stability of the mitotic spindle and chromosomal instability [52,53,54], endocytosis [55], apoptosis [56], protein phosphorylation [57], regulation of gene transcription [58,59,60], regulation of extracellular matrix assembly and tight junctions [61,62,63,64], RNA stability [65,66,67], regulation of senescence [68,69], and, in cytokine signaling pathways, mediating DNA damage response and initiating DNA repair [70] or JAK/STAT signaling [71]. These diverse roles of VHL confirm its implication in several crucial cellular processes, including hypoxia-related cellular adaptations, such as hematopoiesis, metabolic adjustments in oxygen deprived environments, and hypoxia-unrelated mechanisms, such as metabolic rewiring, response to oxidative stress, DNA damage, particularly double-stranded breaks, inflammation, and so on, in the absence or presence of reduced oxygen levels.
Due to its involvement in numerous cellular processes, VHL interacts with several proteins, constituting more or less complex molecular relationships (Figure 3). The occurrence of several VHL isoforms further augments the complexity of VHL biological functions. For example, it has been determined that VHL30, but not VHL19, interacts with tumor suppressor ARF (CDKN2A gene) through the acidic disordered N-terminus. VHL19 lacks the first 53 N-terminal amino acids in this region, and does not interact with ARF [72,73]. ARF promotes interaction between VHL30 and PRMT3, an arginine methyltransferase which can monomethylate or asymmetrically dimethylate several proteins, including p53 and 40S ribosomal protein RPS2 [72]. Methylated p53 arginine residues affect p53 response by influencing the specificity of p53 binding to promoters [74]. Furthermore, VHL isoforms directly interact with p53, which competes for binding to the VHL α region with Elongin-C and ATM, an important cell cycle checkpoint kinase, as well as AURKA, another cell cycle kinase, which is involved in microtubule formation and the regulation of centrosomes, spindles and kinetochores, and centrosomal proteins, e.g., CEP86 [75,76,77]. VHL isoforms, particularly VHL19, have been shown to interact with collagen, fibronectin, and enzymes involved in collagen biosynthesis [72].
3. Clinical Presentation of VHL Disease
Clinical presentation varies in different families, and even in probands within the same family. It is rare to find all manifestations of the disease in one patient. In familial cases, up to 50% of patients have only one manifestation of the syndrome [79]. Clinical criteria for the diagnosis of VHL disease are positive family history for hemangioblastomas in the central nervous system (CNS), or retinal hemangioblastomas; renal cell carcinoma (RCC); pheochromocytoma (PCC), or pancreatic tumors or cysts; and some other tumors. The presence of only one manifestation is sufficient for the diagnosis in familial cases. Diagnostic criteria in sporadic cases are the development of two hemangioblastomas in the CNS or retina, or a hemangioblastoma coupled with visceral tumors or cysts [79,80]. VHL disease is associated with high morbidity and mortality, having a median life expectancy of 50 years. The most frequent causes of death are complications of cerebellar hemangioblastomas (53%) and RCC metastases (32%) [79,81].
There are two clinical types of VHL disease based on the occurrence of typical genotypes and frequencies of RCC or PCC occurrence [3,12,33,82]. Patients without PCC are categorized as type 1 VHL disease, whereas patients with PCC are categorized as having type 2 VHL disease. Type 2 is further sub-classified into three subtypes, A, B, and C, based on the presence or absence of hemangioblastomas and RCC. Type 2A and type 2B both include hemangioblastomas, but RCC is present only in type 2B patients. Type 2C, which is rare [9], includes only PCC, with no hemangioblastomas or RCC [1,80]. Some authors classify Chuvash polycythemia as type 3 VHL disease [83].
4. Genetic and Molecular Basis of VHL Disease
The spectra of pathogenic variations in VHL disease is diverse, and the associations between genotypes and phenotypes—the development of specific tumors—are not always predictable [84]. VHL databases, such as VHLdb (http://vhldb.bio.unipd.it/, accessed on 23 December 2021) and the UMD-VHL mutations database (www.umd.be/VHL/, accessed on 23 December 2021), have collected detailed information on over 1600 different pathogenic variations [85]. The COSMIC (https://cancer.sanger.ac.uk/cosmic, accessed on 23 December 2021) database lists more than 1800 entries for genetic variation in VHL gene [86]. Based on the diverse roles VHL exerts in cellular homeostasis in normoxic and hypoxic conditions, the elucidation of the effect of pathogenic variations at the molecular level is a daunting task. Over the years, the efforts of several research groups enabled greater understanding of the molecular mechanisms underlying different types of pathogenic genetic variants.
The most common VHL disease-associated genetic changes include deletions of exons, in-frame insertions and deletions, truncating point mutations, missense mutations, splice-site mutations, and frameshift insertions and deletions, as well as structural variations and even gene fusions [9,85,87,88]. In general, from the perspective of VHL genetic modifications, type 1 is characterized by exonic deletions and truncating mutations, as well as missense mutations, associated with VHL instability and high HIF activity [1,12,88]. In type 2, regardless of the subtype, missense mutations, which have generally been found to retain partial functionality of VHL protein, are predominantly found [1,88]. Furthermore, patients with nonsense or frameshift mutations are at higher risk for the development of RCC and hemangioblastomas. Clinical investigations demonstrated that, in addition to the type of the VHL genetic variant, the disease type is also age dependent, with a penetrance of over 90% by the age of 65 [79].
Studies involving patients with different manifestations of VHL disease, as well as functional studies of genetic aberrations of VHL, have clearly indicated that some aberrations segregate with distinct phenotypes (Table 1). For example, Ong et al. demonstrated that the risk of developing PCC was associated with VHL mutations that change surface amino acids (termed surface mutations), compared to mutations that change amino acids buried deeper within the structure (termed deep mutations), and/or large deletions and truncating (frameshift and nonsense) mutations [9]. Furthermore, the mean age at diagnosis was lower in PCC patients harboring surface mutations than in those carrying other types of mutations in the VHL gene. They also discovered that RCC and retinal hemangioblastoma patients with deleterious mutations were older at the diagnosis of the disease [9]. Hacker et al. determined that p.R167Q and p.D121G type 2B mutations, located in the Elongin-C binding region, probably retained the ability to bind VBC complex proteins Cullin-2 and Elongin-B, as shown in co-immunoprecipitation experiments [89]. Despite the unstable association of proteins that constitute VBC complexes, this research showed that a p.R167Q VHL mutant could ubiquitinylate HIF1α. Buart et al. showed that an R167Q VHL mutation leads to molecular changes related to more pronounced cancer cell stemness and tumor plasticity. VHL-R167Q expressing RCCs are associated with a poor survival [90].
It has been shown that the endothelium of VHL patients is functionally compromised and more susceptible to tumor development [119]. Specific VHL mutations have been associated with defective blood vessel formation. Deletion of VHL alleles and certain type 2B VHL missense mutations resulted in an increased risk for hemangioblastoma and RCC formation. These mutations cause abnormal, often excessive, blood vessel remodeling, and data from a study performed by Arreola et al. suggested that they have different effects on the nature of vascular changes during the development of retinal vasculature [115]. They analyzed embryonic stem cell-derived blood vessels with Vhl−/−, Vhl2B/2B, and WT backgrounds in constructed mouse models with genotypes (i) conditional Vhl-null genotype, (ii) one wild-type Vhl allele and a second mutant Vhl allele with a type 2B p.G518A mutation (equivalent to the VHL p.R167Q protein variation in humans), and (iii) mutant VHL p.G518A allele and conditional deletion of the wild type Vhl allele (mimicking loss of heterozygosity). Non-mutant mouse models were used as controls. The conditional Vhl-null mutation resulted in accelerated arterial vessel maturation, whereas the type 2B Vhl p.G518A mutation caused an increase in vessel-branching complexity and disrupted Notch and Vegf signaling, also demonstrated by increased Vegfa, Hey2, and Notch3 mRNA levels in enriched endothelial cells Vhl2B/2B extracted from embryonic stem (ES) cell cultures. In comparison, the expression profiles of Vegfa and Notch pathway components in Vhl−/− endothelial cells were different, indicating that aberrant Vegfa and Notch signaling pathways in different genetic backgrounds differ, and thus influence the morphological differences in the development of vasculature. Examination of postnatal mouse retinas, obtained at different postnatal development stages, demonstrated that conditional Vhl-null mutation had a profound effect on the reduction of arterial and venous branching in late stages, and very little effect in early stages. Retinal vessels in Vhl heterozygous mice, harboring wild type allele and a type 2B Vhl mutation, showed increased arterial, but not venous, branching, whereas in conditional Vhl homozygous mice, carrying a type 2B Vhl mutation and conditional deletion of second Vhl allele, both arterial and venous branching were observed. Collectively, these results indicated the differential effect of aberrant Vegfa and Notch signaling linked to Vhl missense mutations, or conditional deletion of Vhl, on vascular (dys)morphogenesis [115,120]. These findings could lead to the identification of novel treatment targets in VHL disease, characterized by extensive vascularization due to overproduction of VEGF.
Genotype–phenotype correlations in VHL disease suggest that oxygen-dependent HIF regulation by VHL mutant proteins, as well as HIF-independent VHL functions, modulate the risk of tumor development. It has been established that certain mutations in the VHL gene resulted in a state of pseudohypoxia with elevated levels of HIF proteins, and subsequent activation of HIF-dependent genes, which upregulate angiogenesis, increased cell proliferation and shifted metabolism toward glycolysis, the pentose phosphate pathway, and glutamine-dependent fatty acid biosynthesis, whereas other mutations preferentially affected HIF-independent pathways, without inducing pseudohypoxia [12,91,121]. The HIF signaling pathway is most frequently activated by inactivating mutations of the VHL gene [122]. Abnormally elevated transcriptional activities of the HIF1α and HIF2α genes have been shown to increase tumor survival in solid tumors [123]. Increased hemoglobin concentrations can occasionally occur because of tumor (hyper)production of erythropoietin, as observed in hemangioblastomas, RCC, and PCC [33,122,124,125]. Interestingly, however, despite the fact that elevated HIFs in the background of certain VHL mutations has been associated with erythrocytosis, this condition is not a common feature of VHL disease.
The complexity of phenotype–genotype associations between VHL aberrations and disease is further demonstrated by research data that VHL genetic aberrations follow the so-called continuum model of tumor suppression, which accounts for the zygosity status of genetic change and tissue specificity [112,126]. Indeed, the research performed by Couve et al. indicated that disease phenotype, in the background of specific VHL mutations, can be dependent on the gradient of VHL loss of function, and can show an additive effect in the context of double mutants [112]. Their research resolved intriguing family cases who were classified as having type 2B VHL disease, based on the presence of CNS and retinal hemangioblastomas, RCC, PCC, and pancreatic neuroendocrine tumors. Initially, only heterozygous p.R200W change was found. This change, in hetero- or homozygous form, was firmly associated with normal phenotype and/or erythrocytosis (Chuvash polycythemia), respectively [127,128,129]. Subsequent analyses revealed another change, p.R161Q, located in the same allele together with the p.R200W change, in diseased probands. Protein change p.R161Q was previously associated with type 2A VHL disease, with low risk for the development of RCC. However, the presence of both mutations in the same allele abrogated HIF2α binding, whereas a single p.R161Q mutant showed only partially impaired binding, and p.R200W binding to HIF2α was within the normal range [127,128,129]. Therefore, the simultaneous presence of these two changes in the VHL gene affected the HIF signaling pathway more profoundly and carriers of double mutations were susceptible to type 2B VHL disease.
5. Clinical and Genetic Features of Selected Manifestations of VHL Disease
5.1. Hemangioblastomas in the Central Nervous System (CNS)
Hemangioblastomas of the CNS are the most frequent manifestations in VHL disease. They occur in 60 to 80% of cases and are a presenting lesion in about 40% of cases. The average age of diagnosis is 29 years. Multiple tumors are always associated with VHL disease [79,88]. Most commonly, they are found in the cerebellum, brainstem, and spinal cord. These tumors are benign, and can remain latent for many years. However, tumor growth can result in a mass effect, with variable signs and symptoms, depending on the location and size of the tumor. Hemangioblastomas with cysts tend to grow faster and become symptomatic sooner [130,131]. Patients with cerebellar or brainstem tumors usually present with symptoms of increased intracranial pressure. Ataxia is a common sign of a cerebellar tumor. Tumors in the spinal cord may be associated with neurogenic pain, sensory deficits, proprioceptive changes, paraparesis, and medullar hypertonicity. Paraneoplastic erythrocytosis may be present in some patients due to overproduction of erythropoietin [80].
Hwang et al. examined 55 Korean patients with VHL disease and found that a germline p.E70K mutation was the most frequent in hemangioblastomas of the CNS and retina [97]. They speculated that this change could be a hotspot for Korean patients, due to previous observations that mutations in the β domain that diminish HIFs binding were associated with RCC, CNS, and retinal hemangioblastomas [96,97]. In this cohort of Korean patients, it was associated with single or multiple hemangioblastomas, and was absent in patients with PCC, RCC, and pancreatic cancer [97]. Screening of sporadic CNS hemangioblastoma cases, performed by Catapano et al., identified two patients with heterozygous germline mutations, p.P86L and p.R167W [132]. Interestingly, the patient with a p.P86L mutation developed a retinal hemangioblastoma four years after the treatment of their CNS hemangioblastoma, whereas in the case of a patient with a p.R167W change, a VHL genetic analysis uncovered this change in a close relative who did not have any clinical manifestations. However, all patients were enrolled in VHL-disease surveillance protocol. A large study, that included 533 patients with VHL disease, evaluated the correlation between developing VHL-related tumors and the type of VHL mutations [133]. The researchers found that truncating and missense mutations in non-Elongin-C binding sites conferred a high risk for the development of CNS hemangioblastomas. Hong et al. performed a mutational analysis on 540 patients from 80 unrelated families [94]. They confirmed that patients carrying the same mutation, regardless of their relation, can develop distinct tumor phenotypes, which can be related to age, ethnic background, or individual molecular background. Their analyses revealed missense mutations (e.g., c.194C>T (p.S65L), c.262T>C (p.W88R), c.269A>T (p.N90I), c.349T>G (p.W117G), c.481C>T (p.R161*), c.486C>G (p.C162W), and c.500G>A (p.R167Q)) that were found at greater frequencies than expected in patients with CNS hemangioblastomas, although it should be noted that other tumors were also observed in the same or different patients within groups [94].
5.2. Retinal Hemangioblastomas and Other Ocular Manifestations
Retinal hemangioblastomas are often the first manifestation of VHL disease. The mean age of onset is 25 years, which is the lowest among other clinical features. Incidence rates have been reported to vary from 49% to 85% [134,135,136]. The likelihood of retinal hemangioblastoma development increases with age, with a probability of 80% after the age of 80 [135]. More than half of patients have bilateral lesions, and around one-third of patients have multiple lesions [134,136,137,138,139,140]. Retinal hemangioblastomas range from small capillary abnormalities to larger lesions that have a typical appearance of a reddish nodule with markedly dilated and tortuous afferent and efferent vessels. Larger tumors can lead to retinal edema, hard exudates, and exudative retinal detachment. Fibrotic changes can result in tractional retinal detachment, which may lead to vitreous hemorrhages, the development of neovascular glaucoma, and eventually phthisis bulbi. Visual loss is generally caused by exudation from the tumor or by traction of glial proliferation on the surface of the tumor. Lesions are mostly located in the retinal periphery (85%); less frequently, they are located near or at the optic disc [12,134,136,137,138,139]. A majority of retinal hemangioblastomas grow over time, but some may be stable for a longer periods, or, although rare, can even spontaneously regress [134,141].
Vascular hamartomas located in the superficial retina were described as small, flat, moss fiber-like vascular lesions, without enlarged afferent and efferent vessels [142]. Juxtapapillary fibrovascular membranes and twin vessels have also been described [134,143,144].
Wittstrom et al. tested retinal function in VHL patients with or without retinal hemangioblastomas [145]. Full-field electroretinography showed significantly prolonged implicit times for rod b-wave (originating from Müller cells and bipolar cells) and maximal combined a-wave (originating from photoreceptors) in VHL patients compared to healthy controls. These results suggested widespread retinal disease even in the absence of a retinal hemangioblastoma or other visible retinal lesions. The same study also showed a significant difference in central retinal thickness between VHL patients and controls; VHL patients had thinner central retinas. This retinal thinning could be secondary to an increased apoptosis of retinal cells [145].
Recently, some researchers analyzed retinal microvasculature using non-invasive optical coherence tomography angiography (OCTA) imaging in patients with VHL disease. Lu et al. analyzed images of 67 eyes with a history of VHL disease, and found significant increases in vessel density of both the superficial and deep capillary plexuses in the macula, regardless the presence of a retinal hemangioma [146]. Similarly, Pilotto et al. found macular perfusion impairment in patients with VHL disease, regardless the presence of a retinal hemangioblastoma [147]. In contrast to the study by Lu et al., patients with VHL disease in this study had lower numbers of retinal vessels in the macular area, and the vessels were thinner in diameter compared to healthy controls. These changes were more pronounced in the superficial capillary plexus [147]. Another study by Pilotto et al. showed thinning of the peripapillary retinal nerve fiber layer in VHL patients without a retinal hemangioblastoma [148]. On the other hand, there was no difference in the peripapillary retinal nerve layer thickness between patients with a retinal hemangioblastoma and healthy controls. The researchers hypothesized that the thinning was a consequence of a reduced perfusion of the radial peripapillary capillary plexus, and that a relative increase in the peripapillary retinal nerve fiber layer, in the presence of a retinal hemangioblastoma, may be due to astrocyte proliferation. All OCTA vascular parameters in the peripapillary area were reduced in VHL patients, and there was no difference between eyes with or without a retinal hemangioblastoma [148].
Optic nerve hemangioblastomas in the intraorbital area, or within the intracranial space, have also been found, although they are very rare. For example, 11 cases (5.3%) were found in a series of 406 patients with VHL disease [137]. Overall, approximately 40 cases of optic nerve hemangioblastomas have been reported in the literature [149,150].
Neovascularization of the iris, and corneal neovascularization, are rare, but have been found in patients with VHL disease. In fact, these are complications of a large retinal hemangioblastoma with longstanding exudative or tractional retinal detachment leading to an extensive retinal ischemia. Iris neovascularization may develop into neovascular glaucoma, and corneal neovascularization may lead to corneal perforation. Both complications result in vision loss [135,137,151].
Genotype analysis performed by Dollfus et al. showed that the majority of patients with retinal hemangioblastomas had missense mutations [152]. Nonsense mutations, frameshifts, and splice site mutations were also detected, albeit at lower frequencies. In this study, in the group of patients with no ocular involvement, in addition to missense mutations, nonsense mutations, and frameshifts, they also found large deletions [152]. Their analyses did not show clear correlations between the location of VHL mutation and the phenotype. They observed that the individuals with truncated VHL proteins may have a decreased risk of developing multiple retinal hemangioblastomas in comparison with individuals who have missense mutations in the VHL gene. Furthermore, the individuals with truncated protein forms who developed retinal hemangioblastoma had lower visual morbidity than patients with other types of VHL mutations. Similar findings were presented in the study performed by Hajjaj et al., in which patients with missense mutations had worse prognoses, developed more aggressive lesions with progression-related complications, and were at higher risk for developing multiple retinal hemangioblastomas when compared to the group of patients who had truncating mutations [138]. The individuals with missense mutations had a greater chance to develop lesions in all four quadrants [138]. In a large study encompassing 406 individuals from 199 families, more than half of examinees were diagnosed with retinal hemangioblastomas, and, with the exception of very young patients, the majority also had accompanying RCC or CNS lesions [137]. The comparison of mutation types revealed that patients with larger deletions, encompassing the whole VHL gene, were less likely to present with ocular manifestations, whereas partial deletions and nonsense mutations, as well as missense mutations, often resulted in the development of a retinal hemangioblastoma. They also described unusual cases of retinal neovascularization mimicking diabetic retinal neovascularization, which is presumably one of the presentations within the spectrum of hemangioblastoma formation. Most of these patients (88%) did not have a typical hemangioblastoma present in the affected eye, but there was a hemangioblastoma present in the fellow eye in 76% of patients. Retinal neovascularization was most often found at the optic disc, with an epiretinal membrane [137]. Mettu et al. focused on 412 patients with VHL disease who had missense mutations in the VHL gene [153]. A total of 159 individuals presented with ocular manifestations, defined as history or clinical evidence in at least one eye. The study found that patients with mutations in the α domain of the VHL gene had a higher prevalence of retinal hemangioblastomas compared to individuals with missense mutations in the functional β domain [153]. In addition, these individuals also had a higher risk of developing a juxtapapillary retinal hemangioblastoma, whereas patients with mutations in the β domain more frequently developed peripheral lesions. Overall, in all patients with VHL disease, the most commonly mutated codons of the VHL protein were 98 and 167, followed by codons 78, 117, and 161.
Interestingly, in the following years of research, despite some conflicting results regarding the associations between type and/or location of genetic variations and the severity of the disease emerged, the majority of studies confirmed more frequent involvement of missense mutations and partial deletions in the development of ocular lesion in patients with VHL disease, in contrast to patients who had complete deletion of the VHL gene [6,137,138,139,145].
5.3. Renal Tumors
Benign renal cysts and malignant RCC are present in two-thirds of patients with VHL disease, and RCC is the leading cause of mortality in VHL disease. The mean age of diagnosis of RCC in VHL disease is 39 years. The risk of RCC varies in different subtypes of VHL disease; however, up to 45% of patients develop multiple and bilateral tumors by the age of 60 years [12,88,130].
Cysts are usually multiple, bilateral, and may be precursors of RCCs. RCCs associated with VHL disease are always of the clear-cell subtype. Büscheck et al. found VHL variations in virtually all renal tumor subtypes. They studied the prevalence and clinical significance of VHL mutations and 3p25 deletions in renal tumor subtypes, and found highly variable ratios of VHL mutations and 3p deletions in the different histological subtypes of kidney tumors. Clear-cell RCC showed the highest rates of VHL alterations, exceeding 80% of all cases. In contrast, VHL alterations did not exceed 30% in the other renal subtypes. These results are important for developing treatment strategies, indicating that anti-VHL treatment strategies should not be only limited to patients with clear-cell RCC [154].
Most cases of RCCs are asymptomatic, but flank pain or hematuria may be present at the time of diagnosis. Renal lesions often grow slowly; therefore, smaller lesions (less than 2 cm) can be clinically followed up. Tumors larger than 4.5 cm are usually associated with metastases [79,80].
It has been firmly established that HIF deregulation is not sufficient in VHL-defective renal carcinogenesis; rather, several additional genetic events are required to cause RCC, including genetic changes in other genes, aberrant epigenetic events, and so on [155,156,157].
Comprehensive molecular characterization of RCC confirmed the association of VHL gene mutations and the involvement of epigenetic reprogramming in the development of the disease [121,158]. PBRM1, or protein polybromo-1, which is a subunit of ATP-dependent chromatin-remodeling complexes, and SET2D, a histone methyltransferase that specifically trimethylates lysine 36 of histone H3 (H3K36me3), were among the top mutated genes in RCC. Interestingly, both genes are located on the short arm of chromosome 3, similar to VHL, in the range of 3p-21 to 3p-25. Interactive network analyses identified 25 sub-networks, among which the subnetwork including VHL and interacting proteins was the most frequently mutated, followed by the PBAF SWI/SNF chromatin remodeling complex, with PBRM1, ARID1A, and SMARCA4 as the key mutated genes, and the PI3K/AKT/MTOR pathway, where PIK3CA and MTOR were the most mutated genes [121]. Comparison of gene expression patterns in patients with VHL mutations or chromatin regulators PBRM1, BAP1, and SETD2 mutations revealed significant differences between the two backgrounds, as well as differences within the backgrounds of distinct chromatin regulators [121].
Another study, in which the researchers sequenced exomes in 79 samples collected from 10 patients, also confirmed the finding that the most mutated genes in RCC were VHL, SET2D, and PBRM1. Notably, their analyses revealed that VHL, and in some cases PRMB1, mutations probably occur early in tumorigenesis. They identified an increase of C>T transitions in the context of CpG sites, and a decrease of A>G transitions [159]. Using exome and whole-genome sequencing of paired tumor and normal samples, Sato et al. additionally validated that VHL, SET2D, and PRBM1 were among the top five mutated genes in RCC [160]. In addition, in approximately 40% of cases, they identified mutations in ELOC (Elongin-C, alias TCEB1), which were linked with the loss of chromosome 8, where ELOC is located. The mutations in ELOC substituted tyrosine at position 79 with either cysteine or serine, in 7 cases, or alanine 100 with proline. Tyr-79 and Ala-100 are essential for Elongin-C binding to VHL protein [160].
5.4. Pheochromocytoma/Paraganglioma
Neuroendocrine tumors, pheochromocytomas (PCCs), and paragangliomas (PGLs), collectively marked as PPGLs, develop from chromaffin cells in the adrenal medulla and parasympathetic or sympathetic ganglia, respectively. In most cases, the tumors are benign, although metastatic disease can develop in approximately 25% tumors [161]. More than one-third and up to 50% of PCCs and PGLs occur as part of inherited syndromes, including multiple endocrine neoplasia type 2 (MEN2), neurofibromatosis type 1, VHL disease, hereditary paraganglioma–pheochromocytoma syndrome, Carney triad, and Carney–Stratakis dyad (National Cancer Institute and [162,163]). PCCs arise in up to 20% of patients with VHL disease. They are usually bilateral, and occur early in life [80]. The mean age at diagnosis is 30 years [9,12]. Patients are often asymptomatic [164]. In contrast to sporadic PCC cases, in many patients with VHL disease there is no excessive catecholamine production by the tumor. However, when there is catecholamine overproduction, some, but not all, patients present with symptoms such as palpitations, sweating, and headache. Undiagnosed catecholamine-producing PCC may result in hypertensive crisis, heart failure, and stroke. Approximately 3% of patients with VHL disease develop a malignant tumor with metastasis [79].
In the context of PPGL molecular taxonomy, genetic aberrations in VHL, EPAS1, succinate dehydrogenase subunits SDHA, SDHB, SDHC, and SDHD, and fumarate hydratase (FH), as well as in PHD1 and PHD2, constitute a pseudohypoxia cluster with specific molecular signatures [162,165,166,167,168]. In VHL-mutated PCC tumor samples, Gao et al. found that seven genes, including CTGF, SDCBP, CYR61/CCN1, COL3A1, COL1A1, COL5A2, and SERPINE1 were significantly up-regulated. This molecular signature proved that VHL mutation could promote the development of PCC by activating the expression of cell proliferation- and migration-associated genes [169]. The second cluster, the kinase signaling cluster, is characterized by mutated genes such as RET, NF1, TMEM127, MAX, HRAS, and KIF1BB, whereas the WNT signaling cluster involves mutations in MAML3 and CSDE1, and is mostly associated with aggressive metastatic sporadic PCCs and PGLs [165,168]. Approximately 70% of PCCs can be explained by sporadic or germline genetic aberrations in these genes [170]. Interestingly, certain mutations in VHL have been related to a high risk of developing PCCs, a low risk of metastasis, and a low risk of developing PGLs [171]. Furthermore, certain germline VHL mutations cause familial PCC without other stigmata of VHL disease (Type 2C VHL disease).
PCCs and low risk PGLs are often bilateral and multicentric, and occur in younger patients with VHL disease, in comparison with other PGL/PCC hereditary syndromes [166,172]. In type 2C VHL disease, which is exclusively associated with the development of PCC, large deletions and truncating mutations are rare, whereas missense mutations, enabling residual VHL function, are more common [1,88,166]. Although the molecular classification of PPGLs included VHL-mutated tumors in pseudohypoxia cluster, several lines of research indicated that variations in the penetrance of different VHL mutations in the development of PCCs within type 2C, and possibly within other VHL 2 type malignancies, might be associated with HIF-independent effects of VHL [1,88]. In particular, the type 2C p.L188V change in the Elongin-C binding domain does not affect its ability to polyubiquitinate HIFs. Type 2C associated changes in the HIF1α binding domain, p.V84L, p.R64P, and p.F119S, also retained the ability to bind HIF1α or had reduced binding ability, respectively, and were able to polyubiquitinate HIF1α [91]. All type 2C mutants did not bind properly to fibronectin, and were able to suppress GLUT1 levels, which indicated that hypoxia-regulated genes were not induced in the context of type 2C typical VHL mutations [91]. The loss of the VHL ability to promote fibronectin matrix assembly could be, therefore, important in type 2C development. Further investigation of VHL mutations in type 2C VHL disease also revealed that all type 2C mutants failed to induce apoptosis in adrenal chromaffin cells through inability to stabilize p53 protein and maintain its activity, whereas HIF1α and HIF2α protein levels were not affected [170]. Type 2C mutations p.R64P, p.F119S, and p.L188V have also been shown to interfere with regulation of transcription factor AP-1 (JUN)-induced apoptosis. VHL, harboring these changes, failed to reduce transcription factor jun-B levels, and failed to polyubiquitinate atypical protein kinase C family members (aPKC) [92]. Increased jun-B levels have been associated with the suppression of apoptosis in an in vitro rat PCC cell line after removal of nerve growth factor (NGF) [92].
On the other hand, investigation of sporadic and familial cases of PCC identified an exon 2-skipping synonymous genetic variant, c.414A>G, p.Pro138Pro (p.P138P), which produced a shorter VHL transcript lacking the HIF binding domain [173]. In five families with type 2A or 2C VHL disease, all affected individuals had PCCs, and type 2A patients also presented with hemangioblastomas, but no RCC. Interestingly, the affected patients were predominantly male. Analysis of TCGA pan-cancer set identified another synonymous variant, p.L188L, which also probably influenced exon 2 exclusion [173]. The complex pattern of VHL splicing in the milieu of genetic changes in VHL exon and intron regions has been previously demonstrated in patients with VHL disease and erythrocytosis [15]. They observed skipping of exon 2 in investigated LCLs (lymphoblastoid cell lines, established from patients) and PCCs from patients carrying a p.P138P heterozygous change with concomitant loss of the wild-type allele, as well as in patients carrying two heterozygous changes in cryptic exon E′ (c.340+617C>G and c.340+648T>C), but with the wild-type VHL allele present. The affected family members in this family were diagnosed either with PCC alone, retinal hemangioblastoma alone, or a combination of PCC, RCC and retinal hemangioblastoma, and CNS hemangioblastoma [15]. Quantification of VHL mRNA from samples (LCLs and PCCs) obtained from these patients showed a higher prevalence of transcripts lacking exon 2, and a higher prevalence of transcripts containing E′ in comparison with controls; however, transcripts containing all three exons were still present. Immunoblot analysis subsequently showed that the protein isoform containing E1 amino acids was absent, whereas isoforms VHL213, VHL172, and VHL160 were underrepresented. The analyses of expression using puromycin (inhibitor of nonsense-mediated mRNA decay, NMD) confirmed that the presence of a stop codon in retained E1′ could activate mRNA degradation via NMD [15]. Buffet et al. studied the incidence of germline mutations in the cryptic exon (E1′) of VHL gene in patients with VHL disease, and in patients with PCC or PGL. They demonstrated that E′ VHL variants are rare, but nearly as frequent as the VHL variants in exons 1 and 2 in patients with PCC or PGL [174]. Hergovich et al. discovered that VHL—specifically cytosolic VHL30—interacts with microtubules in vivo, and that amino acid residues 95–123 are necessary for microtubule stabilization and binding [98]. Several disease-associated genetic variations have been located in this region. After performing a series of experiments utilizing wild-type VHL30 and VHL30 carrying selected missense genetic changes, the researchers concluded that altered microtubule stabilization due to the investigated missense genetic changes, p.Y98H and p.Y112H, contributed predominantly to the development of VHL hemangioblastoma and PCC (classified as type 2A VHL disease), whereas p.F119S-induced destabilization of microtubules was characteristic of the type 2C PCC phenotype [98]. Interestingly, alternative substitutions at positions 98, 112, and 119 (p.Y98N, p.Y112N, and p.F119L, respectively), which are associated with type 2B VHL disease, did not show impaired binding to microtubules in vitro. This study indicated that certain substitutions at specific positions within the VHL microtubule-binding region could either destabilize or stabilize microtubules; furthermore, it appears that microtubule stabilization, characteristic of certain type 2B VHL variants, could be incompatible with RCC development [98].
6. Case Report—Patient with VHL Disease
A thirty-three year old female began to have severe headaches 14 days before presentation. The headaches were mostly located occipitally, and were more severe in the mornings. One day before presentation, the patient noticed transient left hemifacial tingling. Her past medical history and family history were negative. Neurologic examination revealed mild left upper limb ataxia, dysdiadochokinesis, and right extensor plantar response. Mildly impaired vibration sense was found in the lower limbs. No other abnormalities were found. Romberg’s sign was negative.
The patient underwent extensive diagnostics. Magnetic resonance imaging (MRI) of the head showed a 45 × 20 mm large, partly cystic space-occupying lesion at the base of the 4th ventricle, pressing the brainstem and upper spinal cord (Figure 4A). Two similar lesions were present in the left cerebellar hemisphere (Figure 4B). There were several similar lesions evident in the spinal cord MRI, the largest one being located at the C2 level, and the others at the C5-C6, Th1-Th3, Th5, and Th10 levels (Figure 4C). All lesions were consistent with hemangioblastomas.
Two small peripheral retinal hemangioblastomas, some small telangiectatic changes, and a peripheral gliotic lesion with a vitreous traction were found on ophthalmological examination (Figure 5).
Abdominal MRI revealed multiple hepatic and pancreatic cysts and a small cortical cyst in the left kidney. Endocrinological examination ruled out the presence of PCC. The clinical diagnosis of VHL disease was confirmed with genetic testing. A known pathogenic missense mutation, VHL c.194C>T (p.S65L), was found.
Patient first underwent a surgical procedure for the removal of a life-threatening expansive tumor near the 4th ventricle. Histological examination of the excised tumor confirmed the radiological diagnosis of hemangioblastoma (WHO grade I). Brainstem and cerebellar tumors, as well as a spinal cord tumor at the Th5 level, were removed in two additional surgical procedures in the following year. Retinal hemangioblastomas were successfully treated with laser photocoagulation. Since then, the patient has had regular check-ups. In the following three years there was no significant progression of the disease.
Commentary on the Patient’s Genotype–Phenotype Correlation
Takayanagi et al. identified a c.194C>T transition in VHL-related cerebellum hemangioblastoma, and a nonsense transversion, c.194C>A (p.S65*), in a sporadic case of cerebellum hemangioblastoma, which also harbored additional VHL mutations, c.458T>G and VHL promoter methylation, resulting in biallelic inactivation of VHL [175]. Using different in vitro approaches, Miller et al. investigated VHL codon 65 substitutions p.S65L, p.S65W, and p.S65A [95]. Protein changes p.S65L and p.S65W were previously identified in angiomas and angiomas/hemangioblastomas, respectively [176,177]. All three mutant VHL proteins retained binding capacity for Elongin-B/C, as expected; however, both p.S65L and p.S65W did not bind hydroxylated HIF1α, and were unable to induce HIF2α (EPAS1) degradation. The 65 codon is located in the linker L1 loop, and computational analyses of the substitution of serine with lysine showed that serine plays an important role in the binding of HIF1α to the β domain of VHL, and is possibly also responsible for the dynamic coupling of HIF1α–β domain to the Elongin-C-α domain [95]. A large study, performed on 540 patients, identified a c194C>T mutation, predominantly in CNS hemangioblastomas, followed by RCC, pancreatic tumors/cysts, the genital system (epididymis or broad ligament), retinal hemangioblastoma, and PCC [94].
Determining a genetic cause of the disease presents a good template for establishing optimal clinical management of the patient and genetic counseling for family members.
7. Erythrocytosis in the Context of VHL Genetic Changes
Erythrocytosis is defined as an abnormally increased red blood cell count [178]. Its occurrence may be seen in various conditions. Interestingly, there are many conditions associated with erythrocytosis, in which erythrocytosis is presumably caused by chronic HIF activation, and yet there are no tumors present in these conditions. Erythrocytosis has been associated with cardiovascular morbidity [179]. According to the retrospective analysis performed by Nguyen at al., the rates of thromboembolic events prior to diagnosis were comparable in both polycythemia vera and secondary erythrocytosis patients. Three out of one hundred and two patients experienced venous thrombosis; all three of these patients were diagnosed with polycythemia vera [180]. Changes in blood composition disturb microvascular circulation and lead to hyperviscosity and thrombotic events. Ophthalmological examination of the fundus may reveal venous dilatation and tortuosity, intraretinal hemorrhages, retinal edema, hard exudates, cotton wool spots, and optic disc edema. Although ocular changes have been well documented in patients with polycythemia vera [181,182,183,184], and may occur early in the progression of the disease, the occurrence of ocular lesions in other types of erythrocytosis is less well documented.
It has been established that the inheritance of VHL genetic changes is an important cause of secondary erythrocytosis [185]. Up to 50% of patients with apparent congenital erythrocytosis and elevated serum EPO appear to have a mutation in the VHL gene [186]. The OMIM (https://omim.org/, accessed on 24 December 2021) database categorizes VHL-induced erythrocytosis as familial erythrocytosis ECYT2, displaying features of both primary and secondary erythrocytosis, such as hypersensitivity of erythroid progenitors to circulating EPO, as well as raised blood EPO levels [187].
Chuvash polycythemia, first observed in the Russian Chuvash population and associated with a p.R200W change in the VHL gene [127,128,129], was later found in patients of other ethnic groups [124,186,188]. Subsequent genetic analyses revealed several other missense homozygous and compound heterozygous genetic changes in VHL as the underlying cause of this disease [186]. Gordeuk et al. analyzed vascular complications among patients with Chuvash polycythemia and found a positive history of major arterial or venous thrombosis in 24% of patients, compared to only 4% of controls. In addition, a history of bleeding was found in 32% of patients with Chuvash polycythemia, compared to 4% of controls. An increased risk for thrombosis was associated with homozygosity for the 598C>T (p.R200W) VHL change. Interestingly, patients with Chuvash polycythemia had no CNS hemangioblastomas, RCCs, or PCCs [124,189]. It has been established that quantitative differences with respect to HIF, and qualitative differences with respect to HIF-independent VHL functions, probably account for the low renal cancer risk associated with Chuvash polycythemia and the high renal carcinoma risk associated with complete loss of function of VHL in renal cells [112,125]. In addition to ECYT2, caused by homozygous (or compound heterozygous) hypomorphic VHL alleles, secondary erythrocytosis can result from genetic changes in other genes in the oxygen-sensing pathway, such as EPAS1 (HIF2α protein) alleles (ECYT4) or EGLN1 (PHD2 protein) alleles (ECYT3). This suggests that subtle defects in the VHL-PHD2-HIF2α pathway can cause polycythemia without significant increase of carcinogenesis [125,190].
Pastore et al. suggested there is a distinction between “polycythemia-causing” VHL variations and other VHL variations causing VHL syndrome with tumor development [186]. They investigated patients with polycythemia from different ethnic backgrounds and found homozygous and compound heterozygous changes, including 598C>T (p.R200W), 571C>G (p.H191D), 562C>G (p.L188V), and 574C>T (p.P192A). They concluded that compound heterozygous changes, such as P192A and L188V in one allele and “polycythemia-causing” p.R200W in the second allele, could be termed “polycythemia associated” VHL genetic changes. These changes are located in the C-terminal region, at the end of the Elongin-C binding region, or just outside it. It has been shown that L188V affected VHL–fibronectin interaction in the context of RCC and PCC, and did not impair HIF1A degradation [91,186]. They speculated that it is possible that the newly identified changes in VHL also do not greatly affect VHL function in HIF1α regulation [186]. In a functional investigation of p.R200W change, the researchers identified enhanced p.R200W and p.H191D VHL interaction with SOCS1, which interfered with JAK2 recruitment and subsequent degradation of JAK2 [71]. The resulting JAK2 stabilization and JAK2-STAT5 stimulation of erythroid progenitors in these patients could, in part, explain hypersensitivity to EPO, which is characteristic of primary erythrocytosis [71]. These findings could be exploited further to investigate whether patients with Chuvash polycythemia would respond to pharmacologic JAK2 inhibition.
However, further research of the homozygous change p.H191D, which was present in one male patient with elevated EPO from Croatia [186], and subsequently confirmed in another Croatian female patient with elevated EPO [191], uncovered the possibility that different mechanisms could drive pathogenic characteristics in carriers of p.R200W (Chuvash) and p.H191D (Croatian) variants. The authors observed that EPO concentrations in both patients with the p.H191D variant were higher than in carriers of a homozygous p.R200W change. Native erythroid progenitors of p.H191D carriers were not hypersensitive to EPO, indicating that this phenotype could be driven exclusively by EPO [191].
Next, despite initial observations that Chuvash polycythemia and congenital erythrocytosis VHL genetic changes clustered in exon 3, it has been found that different changes in exon 2 could also underlie the development of erythrocytosis, without malignant manifestations characteristic of VHL changes in other exons. For example, the discovery and investigation of another homozygous variant, p.D126N in exon 2, in one pediatric patient with early onset pulmonary hypertension, polycythemia, and multiple hepatic hemangiomas, also indicated only mild EPO hypersensitivity of erythroid progenitors. Expression analyses showed normal levels of NF-E2 and RUNX1 mRNA, which are associated with EPO hypersensitivity in primary and Chuvash (p.R200W) polycythemias [192,193]. This patient had remarkably high EPO concentrations—higher than those present in patients with Chuvash (p.R200W) or Croatian (p.H191D) variants [193].
Lanikova et al. identified a homozygous exon 2 VHL P138L variant in a patient with congenital polycythemia [194]. Heterozygous relatives did not present any cancerous or non-cancerous manifestations of VHL disease. Functional analyses showed an association between p.P138L and an increase in the expression of HIF-regulated genes SLC2A1, TFRC, and HK, as well as RUNX1/AML1 and NF-E2, which are increased in acquired and primary polycythemia, respectively. The mutated VHL protein also showed reduced stability and decreased ability to polyubiquitinate HIF1α [194]. Lenglet et al. used several approaches to discern the functional effects of VHL genetic changes in patients with erythrocytosis and VHL disease [15]. They demonstrated that synonymous heterozygous mutation in patients with erythrocytosis induced the expression of an additional VHL isoform, a 193 amino acid protein containing the first 114 amino acids of exon 1 and an additional 79 amino acids spliced from cryptic exon E1′. They also identified polymorphic sites in E′ (c.340+1770T>C, c.340+1694_711dup, and c.340+1816A>C) in patients with erythrocytosis, constituting complex compound genotypes in these patients. Similar to the previously mentioned observation in PCC patients with distinct VHL variations, expression studies of these variations also indicated that, despite higher mRNA levels of transcripts containing E′, they could not detect E′-containing VHL isoforms, indicating that these transcripts may be targeted for NMD. A synonymous homozygous p.D143D change in patients with erythrocytosis was associated with exon 2 skipping, resulting in elevated levels of transcripts containing only exons 1 and 3; however, western blot analysis showed that all VHL isoforms were downregulated. Further analysis of variants, associated with erythrocytosis, p.G144R, and p.P138L, as well as a variant, p.P138P, which was associated with VHL disease, revealed that their effect on splicing is differential, with most splicing events occurring in p.P138P background [15].
In another study, investigation of cases and relatives with unexplained erythrocytosis revealed that 61 of 84 patients with suspected congenital erythrocytosis were carriers of an intron genetic variant, c.-195G>A [195]. Further screening of 78 probands and their relatives indicated that AA homozygotes and GA heterozygotes were significantly overrepresented in patients’ group. This research indicates that idiopathic erythrocytosis could be explained in the background of common single genetic variations (SNVs), possibly in interaction with other SNVs in VHL or VHL binding partners.
8. Conclusions
Hereditary genetic changes in the VHL gene that result in the development of different tumors and erythrocytosis show great complexity, and present a significant challenge with regard to the elucidation of variants association with phenotypes. Recent research has revealed the involvement of VHL in different cellular processes and molecular pathways, both HIF-dependent and HIF-independent, as well as its ability to interact with many different proteins. Therefore, it is expected that the pathogenic effects of VHL variants will influence different mechanisms in cells. The association of VHL variants located at different positions, and the association of different amino acid substitutions at the same position, with distinct phenotypes, could, in the future, aid in the uncovering of pathogenic mechanisms, leading to the development of particular phenotypes of the disease. The identification of VHL-interacting protein partners could further shed light on molecular pathways involved in the initiation and progression of VHL disease. This, in turn, could lead to the development of novel therapeutic approaches and the possibility of optimizing follow-up procedures for carriers of VHL mutations. In addition, the finding that synonymous genetic changes of VHL can influence mRNA splicing mechanisms is important, not only for elucidation of VHL disease mechanisms, but also for reconsideration of synonymous changes and their possible deleterious effects on mRNA maturation in other pathological conditions.
Nevertheless, besides family history and clinical presentation, molecular genetic testing of VHL variations is becoming an important tool in confirming the diagnosis. Genetic testing proved to be indispensable in patients with unclear family history. Understanding the pathogenic effects of VHL variants might better predict the prognosis and help medical professionals to optimize the management of the patient.
Author Contributions
Conceptualization, M.U.; methodology, M.U. and P.H.; investigation, M.U. and P.H.; resources, M.U. and P.H.; writing—original draft preparation, M.U. and P.H.; writing—review and editing, M.U. and P.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Slovenian Research Agency grant number L3-9279.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Written consent has been obtained from the patient for the publication.
Data Availability Statement
In preparation of the manuscript, the following publicly available datasets were used: Human Protein Atlas, version 20.1 (https://www.proteinatlas.org/) (accessed on 10 January 2022), STRING (https://string-db.org/) (accessed on 14 January 2022), VHLdb (http://vhldb.bio.unipd.it/) (accessed on 23 December 2021), UMD- VHL mutations database (www.umd.be/VHL/) (accessed on 23 December 2021), COSMIC (https://cancer.sanger.ac.uk/cosmic) (accessed on 23 December 2021), NCBI RefSeq (https://www.ncbi.nlm.nih.gov/refseq/) (accessed on 24 December 2021), cBioPortal (https://www.cbioportal.org/) (accessed on 30 December 2021).
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chou, A.; Toon, C.; Pickett, J.; Gill, A.J. von Hippel-Lindau syndrome. Front. Horm. Res. 2013, 41, 30–49. [Google Scholar] [CrossRef] [PubMed]
- Chittiboina, P.; Lonser, R.R. Von Hippel-Lindau disease. Handb. Clin. Neurol. 2015, 132, 139–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. von Hippel-Lindau disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, C.; Frerich, J.M.; Germanwala, A.; Yang, C.; Lonser, R.R.; Mao, Y.; Zhuang, Z.; Zhang, M. De novo VHL germline mutation detected in a patient with mild clinical phenotype of von Hippel-Lindau disease. J. Neurosurg. 2014, 121, 384–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasker, S.; Sohn, T.S.; Okamoto, H.; Li, J.; Lonser, R.R.; Oldfield, E.H.; Vortmeyer, A.O.; Zhuang, Z. Second hit deletion size in von Hippel-Lindau disease. Ann. Neurol. 2006, 59, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Murro, V.; Lippera, M.; Mucciolo, D.P.; Canu, L.; Ercolino, T.; De Filpo, G.; Giorgio, D.; Traficante, G.; Sodi, A.; Virgili, G.; et al. Outcome and genetic analysis of patients affected by retinal capillary hemangioblastoma in von Hippel Lindau syndrome. Mol. Vis. 2021, 27, 542–554. [Google Scholar]
- Richards, F.M.; Payne, S.J.; Zbar, B.; Affara, N.A.; Ferguson-Smith, M.A.; Maher, E.R. Molecular analysis of de novo germline mutations in the von Hippel-Lindau disease gene. Hum. Mol. Genet. 1995, 4, 2139–2143. [Google Scholar] [CrossRef]
- Ang, S.O.; Chen, H.; Hirota, K.; Gordeuk, V.R.; Jelinek, J.; Guan, Y.; Liu, E.; Sergueeva, A.I.; Miasnikova, G.Y.; Mole, D.; et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002, 32, 614–621. [Google Scholar] [CrossRef]
- Ong, K.R.; Woodward, E.R.; Killick, P.; Lim, C.; Macdonald, F.; Maher, E.R. Genotype-phenotype correlations in von Hippel-Lindau disease. Hum. Mutat. 2007, 28, 143–149. [Google Scholar] [CrossRef]
- Woodward, E.R.; Eng, C.; McMahon, R.; Voutilainen, R.; Affara, N.A.; Ponder, B.A.; Maher, E.R. Genetic predisposition to phaeochromocytoma: Analysis of candidate genes GDNF, RET and VHL. Hum. Mol. Genet. 1997, 6, 1051–1056. [Google Scholar] [CrossRef] [Green Version]
- Bento, C.; Percy, M.J.; Gardie, B.; Maia, T.M.; van Wijk, R.; Perrotta, S.; Della Ragione, F.; Almeida, H.; Rossi, C.; Girodon, F.; et al. Genetic basis of congenital erythrocytosis: Mutation update and online databases. Hum. Mutat. 2014, 35, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Aronow, M.E.; Wiley, H.E.; Gaudric, A.; Krivosic, V.; Gorin, M.B.; Shields, C.L.; Shields, J.A.; Jonasch, E.W.; Singh, A.D.; Chew, E.Y. VON HIPPEL-LINDAU DISEASE: Update on Pathogenesis and Systemic Aspects. Retina 2019, 39, 2243–2253. [Google Scholar] [CrossRef] [PubMed]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Blankenship, C.; Naglich, J.G.; Whaley, J.M.; Seizinger, B.; Kley, N. Alternate choice of initiation codon produces a biologically active product of the von Hippel Lindau gene with tumor suppressor activity. Oncogene 1999, 18, 1529–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenglet, M.; Robriquet, F.; Schwarz, K.; Camps, C.; Couturier, A.; Hoogewijs, D.; Buffet, A.; Knight, S.J.L.; Gad, S.; Couve, S.; et al. Identification of a new VHL exon and complex splicing alterations in familial erythrocytosis or von Hippel-Lindau disease. Blood 2018, 132, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliopoulos, O.; Ohh, M.; Kaelin, W.G., Jr. pVHL19 is a biologically active product of the von Hippel-Lindau gene arising from internal translation initiation. Proc. Natl. Acad. Sci. USA 1998, 95, 11661–11666. [Google Scholar] [CrossRef] [Green Version]
- Richards, F.M.; Schofield, P.N.; Fleming, S.; Maher, E.R. Expression of the von Hippel-Lindau disease tumour suppressor gene during human embryogenesis. Hum. Mol. Genet. 1996, 5, 639–644. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, A.; Davidowitz, E.J.; Burk, R.D. A second major native von Hippel-Lindau gene product, initiated from an internal translation start site, functions as a tumor suppressor. Proc. Natl. Acad. Sci. USA 1998, 95, 8817–8822. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Min, J.H.; Yang, H.; Ivan, M.; Gertler, F.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of an HIF-1alpha -pVHL complex: Hydroxyproline recognition in signaling. Science 2002, 296, 1886–1889. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, C.E.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of the VHL-ElonginC-ElonginB complex: Implications for VHL tumor suppressor function. Science 1999, 284, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, E.; Murgia, A.; Tosatto, S.C. Adding structural information to the von Hippel-Lindau (VHL) tumor suppressor interaction network. FEBS Lett. 2009, 583, 3704–3710. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H.; Hirai, S.; Takaki, Y.; Kamada, M.; Baba, M.; Sakai, N.; Kishida, T.; Kaneko, S.; Yao, M.; Ohno, S.; et al. Direct interaction of the β-domain of VHL tumor suppressor protein with the regulatory domain of atypical PKC isotypes. Biochem. Biophys. Res. Commun. 1999, 263, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallstrom, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.H.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef]
- Perl, K.; Ushakov, K.; Pozniak, Y.; Yizhar-Barnea, O.; Bhonker, Y.; Shivatzki, S.; Geiger, T.; Avraham, K.B.; Shamir, R. Reduced changes in protein compared to mRNA levels across non-proliferating tissues. BMC Genom. 2017, 18, 305. [Google Scholar] [CrossRef]
- Hsu, T. Complex cellular functions of the von Hippel-Lindau tumor suppressor gene: Insights from model organisms. Oncogene 2012, 31, 2247–2257. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Mao, C.; Wang, X.; Shi, Y.; Tao, Y. Epigenetic crosstalk between hypoxia and tumor driven by HIF regulation. J. Exp. Clin. Cancer Res. 2020, 39, 224. [Google Scholar] [CrossRef]
- Liao, C.; Zhang, Q. Understanding the Oxygen-Sensing Pathway and Its Therapeutic Implications in Diseases. Am. J. Pathol. 2020, 190, 1584–1595. [Google Scholar] [CrossRef] [PubMed]
- Tomc, J.; Debeljak, N. Molecular Insights into the Oxygen-Sensing Pathway and Erythropoietin Expression Regulation in Erythropoiesis. Int. J. Mol. Sci. 2021, 22, 7074. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, O.; Kibel, A.; Gray, S.; Kaelin, W.G., Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat. Med. 1995, 1, 822–826. [Google Scholar] [CrossRef]
- Kibel, A.; Iliopoulos, O.; DeCaprio, J.A.; Kaelin, W.G., Jr. Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science 1995, 269, 1444–1446. [Google Scholar] [CrossRef]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Iliopoulos, O.; Levy, A.P.; Jiang, C.; Kaelin, W.G., Jr.; Goldberg, M.A. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10595–10599. [Google Scholar] [CrossRef] [Green Version]
- Lonergan, K.M.; Iliopoulos, O.; Ohh, M.; Kamura, T.; Conaway, R.C.; Conaway, J.W.; Kaelin, W.G., Jr. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol. Cell Biol. 1998, 18, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzen, E.; Ratcliffe, P.J. HIF hydroxylation and cellular oxygen sensing. Biol. Chem. 2004, 385, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Hirsila, M.; Koivunen, P.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003, 278, 30772–30780. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.A.; Sutphin, P.D.; Yen, S.E.; Giaccia, A.J. Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 α. Mol. Cell Biol. 2005, 25, 6415–6426. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.M.; Yeoh, K.K.; Lee, M.K.; Eriksson, T.; Kessler, B.M.; Kramer, H.B.; Edelmann, M.J.; Willam, C.; Pugh, C.W.; Schofield, C.J.; et al. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J. Biol. Chem. 2011, 286, 13041–13051. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Batty-Stuart, S.; Lee, J.E.; Ohh, M. HIF-1alpha Hydroxyprolines Modulate Oxygen-Dependent Protein Stability Via Single VHL Interface With Comparable Effect on Ubiquitination Rate. J. Mol. Biol. 2021, 433, 167244. [Google Scholar] [CrossRef]
- Hewitson, K.S.; McNeill, L.A.; Riordan, M.V.; Tian, Y.M.; Bullock, A.N.; Welford, R.W.; Elkins, J.M.; Oldham, N.J.; Bhattacharya, S.; Gleadle, J.M.; et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J. Biol. Chem. 2002, 277, 26351–26355. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Zhou, M.I.; Wang, H.; Ross, J.J.; Kuzmin, I.; Xu, C.; Cohen, H.T. The von Hippel-Lindau tumor suppressor stabilizes novel plant homeodomain protein Jade-1. J. Biol. Chem. 2002, 277, 39887–39898. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Bai, M.; Mittal, A.K.; El-Jouni, W.; Zhou, J.; Cohen, D.M.; Zhou, M.I.; Cohen, H.T. Candidate tumor suppressor and pVHL partner Jade-1 binds and inhibits AKT in renal cell carcinoma. Cancer Res. 2013, 73, 5371–5380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermer, B.; Ghenoiu, C.; Bartram, M.; Muller, R.U.; Kotsis, F.; Hohne, M.; Kuhn, W.; Rapka, M.; Nitschke, R.; Zentgraf, H.; et al. The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J. Cell Biol. 2006, 175, 547–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frew, I.J.; Smole, Z.; Thoma, C.R.; Krek, W. Genetic deletion of the long isoform of the von Hippel-Lindau tumour suppressor gene product alters microtubule dynamics. Eur. J. Cancer 2013, 49, 2433–2440. [Google Scholar] [CrossRef] [PubMed]
- Thoma, C.R.; Toso, A.; Gutbrodt, K.L.; Reggi, S.P.; Frew, I.J.; Schraml, P.; Hergovich, A.; Moch, H.; Meraldi, P.; Krek, W. VHL loss causes spindle misorientation and chromosome instability. Nat. Cell Biol. 2009, 11, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.; Adereth, Y.; Kose, N.; Dammai, V. Endocytic function of von Hippel-Lindau tumor suppressor protein regulates surface localization of fibroblast growth factor receptor 1 and cell motility. J. Biol. Chem. 2006, 281, 12069–12080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinesh, G.G.; Kamat, A.M. RalBP1 and p19-VHL play an oncogenic role, and p30-VHL plays a tumor suppressor role during the blebbishield emergency program. Cell Death Discov. 2017, 3, 17023. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Minamishima, Y.A.; Yan, Q.; Schlisio, S.; Ebert, B.L.; Zhang, X.; Zhang, L.; Kim, W.Y.; Olumi, A.F.; Kaelin, W.G., Jr. pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF-kappaB agonist Card9 by CK2. Mol. Cell. 2007, 28, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsova, A.V.; Meller, J.; Schnell, P.O.; Nash, J.A.; Ignacak, M.L.; Sanchez, Y.; Conaway, J.W.; Conaway, R.C.; Czyzyk-Krzeska, M.F. von Hippel-Lindau protein binds hyperphosphorylated large subunit of RNA polymerase II through a proline hydroxylation motif and targets it for ubiquitination. Proc. Natl. Acad. Sci. USA 2003, 100, 2706–2711. [Google Scholar] [CrossRef] [Green Version]
- Mikhaylova, O.; Ignacak, M.L.; Barankiewicz, T.J.; Harbaugh, S.V.; Yi, Y.; Maxwell, P.H.; Schneider, M.; Van Geyte, K.; Carmeliet, P.; Revelo, M.P.; et al. The von Hippel-Lindau tumor suppressor protein and Egl-9-Type proline hydroxylases regulate the large subunit of RNA polymerase II in response to oxidative stress. Mol. Cell Biol. 2008, 28, 2701–2717. [Google Scholar] [CrossRef] [Green Version]
- Na, X.; Duan, H.O.; Messing, E.M.; Schoen, S.R.; Ryan, C.K.; di Sant’Agnese, P.A.; Golemis, E.A.; Wu, G. Identification of the RNA polymerase II subunit hsRPB7 as a novel target of the von Hippel-Lindau protein. EMBO J. 2003, 22, 4249–4259. [Google Scholar] [CrossRef] [Green Version]
- Calzada, M.J.; Esteban, M.A.; Feijoo-Cuaresma, M.; Castellanos, M.C.; Naranjo-Suarez, S.; Temes, E.; Mendez, F.; Yanez-Mo, M.; Ohh, M.; Landazuri, M.O. von Hippel-Lindau tumor suppressor protein regulates the assembly of intercellular junctions in renal cancer cells through hypoxia-inducible factor-independent mechanisms. Cancer Res. 2006, 66, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Ohh, M.; Yauch, R.L.; Lonergan, K.M.; Whaley, J.M.; Stemmer-Rachamimov, A.O.; Louis, D.N.; Gavin, B.J.; Kley, N.; Kaelin, W.G., Jr.; Iliopoulos, O. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol. Cell 1998, 1, 959–968. [Google Scholar] [CrossRef]
- Sevilla-Montero, J.; Bienes-Martinez, R.; Labrousse-Arias, D.; Fuertes-Yebra, E.; Ordonez, A.; Calzada, M.J. pVHL-mediated regulation of the anti-angiogenic protein thrombospondin-1 decreases migration of Clear Cell Renal Carcinoma Cell Lines. Sci. Rep. 2020, 10, 1175. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Mack, F.; Haase, V.H.; Simon, M.C.; Johnson, R.S. pVHL function is essential for endothelial extracellular matrix deposition. Mol. Cell Biol. 2006, 26, 2519–2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danilin, S.; Sourbier, C.; Thomas, L.; Rothhut, S.; Lindner, V.; Helwig, J.J.; Jacqmin, D.; Lang, H.; Massfelder, T. von Hippel-Lindau tumor suppressor gene-dependent mRNA stabilization of the survival factor parathyroid hormone-related protein in human renal cell carcinoma by the RNA-binding protein HuR. Carcinogenesis 2009, 30, 387–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galban, S.; Martindale, J.L.; Mazan-Mamczarz, K.; Lopez de Silanes, I.; Fan, J.; Wang, W.; Decker, J.; Gorospe, M. Influence of the RNA-binding protein HuR in pVHL-regulated p53 expression in renal carcinoma cells. Mol. Cell Biol. 2003, 23, 7083–7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, H.; Brown, J.A.; Gong, C.; Fan, H.; Brewer, G.; Gnarra, J.R. Association of the von Hippel-Lindau protein with AUF1 and posttranscriptional regulation of VEGFA mRNA. Mol. Cancer Res. 2012, 10, 108–120. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.Y.; Sharpless, N.E. VHL inactivation: A new road to senescence. Cancer Cell 2008, 13, 295–297. [Google Scholar] [CrossRef] [Green Version]
- Young, A.P.; Schlisio, S.; Minamishima, Y.A.; Zhang, Q.; Li, L.; Grisanzio, C.; Signoretti, S.; Kaelin, W.G., Jr. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat. Cell Biol. 2008, 10, 361–369. [Google Scholar] [CrossRef]
- Metcalf, J.L.; Bradshaw, P.S.; Komosa, M.; Greer, S.N.; Stephen Meyn, M.; Ohh, M. K63-ubiquitylation of VHL by SOCS1 mediates DNA double-strand break repair. Oncogene 2014, 33, 1055–1065. [Google Scholar] [CrossRef]
- Russell, R.C.; Sufan, R.I.; Zhou, B.; Heir, P.; Bunda, S.; Sybingco, S.S.; Greer, S.N.; Roche, O.; Heathcote, S.A.; Chow, V.W.; et al. Loss of JAK2 regulation via a heterodimeric VHL-SOCS1 E3 ubiquitin ligase underlies Chuvash polycythemia. Nat. Med. 2011, 17, 845–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Song, M.; Hakala, K.; Weintraub, S.T.; Shiio, Y. Proteomic dissection of the von Hippel-Lindau (VHL) interactome. J. Proteome Res. 2011, 10, 5175–5182. [Google Scholar] [CrossRef] [Green Version]
- Minervini, G.; Mazzotta, G.M.; Masiero, A.; Sartori, E.; Corra, S.; Potenza, E.; Costa, R.; Tosatto, S.C. Isoform-specific interactions of the von Hippel-Lindau tumor suppressor protein. Sci. Rep. 2015, 5, 12605. [Google Scholar] [CrossRef] [PubMed]
- Jansson, M.; Durant, S.T.; Cho, E.C.; Sheahan, S.; Edelmann, M.; Kessler, B.; La Thangue, N.B. Arginine methylation regulates the p53 response. Nat. Cell Biol. 2008, 10, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.S.; Kim, H.; Lee, S.M.; Kim, S.T.; Cho, E.J.; Youn, H.D. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol. Cell 2006, 22, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Yin, H.; Yuan, L. Centrosomal MCM7 strengthens the Cep68-VHL interaction and excessive MCM7 leads to centrosome splitting resulting from increase in Cep68 ubiquitination and proteasomal degradation. Biochem. Biophys. Res. Commun. 2017, 489, 497–502. [Google Scholar] [CrossRef]
- Hasanov, E.; Chen, G.; Chowdhury, P.; Weldon, J.; Ding, Z.; Jonasch, E.; Sen, S.; Walker, C.L.; Dere, R. Ubiquitination and regulation of AURKA identifies a hypoxia-independent E3 ligase activity of VHL. Oncogene 2017, 36, 3450–3463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Crespigio, J.; Berbel, L.C.L.; Dias, M.A.; Berbel, R.F.; Pereira, S.S.; Pignatelli, D.; Mazzuco, T.L. Von Hippel-Lindau disease: A single gene, several hereditary tumors. J. Endocrinol. Investig. 2018, 41, 21–31. [Google Scholar] [CrossRef]
- Shuin, T.; Yamasaki, I.; Tamura, K.; Okuda, H.; Furihata, M.; Ashida, S. Von Hippel-Lindau disease: Molecular pathological basis, clinical criteria, genetic testing, clinical features of tumors and treatment. Jpn. J. Clin. Oncol. 2006, 36, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Wang, J.; Liu, S.; Peng, X.; Hong, B.; Zhou, J.; Ma, K.; Zhang, J.; Cai, L.; Gong, K. Hemangioblastoma Instead of Renal Cell Carcinoma Plays a Major Role in the Unfavorable Overall Survival of Von Hippel-Lindau Disease Patients. Front. Oncol. 2019, 9, 1037. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Kishida, T.; Yao, M.; Hustad, T.; Glavac, D.; Dean, M.; Gnarra, J.R.; Orcutt, M.L.; Duh, F.M.; Glenn, G.; et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: Correlations with phenotype. Hum. Mutat. 1995, 5, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Coco, D.; Leanza, S. Von Hippel-Lindau Syndrome: Medical Syndrome or Surgical Syndrome? A Surgical Perspective. J. Kidney Cancer VHL 2022, 9, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kim, K.J.; Hong, N.; Shin, S.; Choi, J.R.; Kang, S.W.; Lee, S.T.; Rhee, Y. Genetic Analysis and Clinical Characteristics of Hereditary Pheochromocytoma and Paraganglioma Syndrome in Korean Population. Endocrinol. Metab. 2020, 35, 858–872. [Google Scholar] [CrossRef]
- Tabaro, F.; Minervini, G.; Sundus, F.; Quaglia, F.; Leonardi, E.; Piovesan, D.; Tosatto, S.C. VHLdb: A database of von Hippel-Lindau protein interactors and mutations. Sci. Rep. 2016, 6, 31128. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Maher, E.R.; Sandford, R.N. von Hippel-Lindau Disease: An Update. Curr. Genet. Med. Rep. 2019, 7, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Hacker, K.E.; Lee, C.M.; Rathmell, W.K. VHL type 2B mutations retain VBC complex form and function. PLoS ONE 2008, 3, e3801. [Google Scholar] [CrossRef] [Green Version]
- Buart, S.; Terry, S.; Diop, M.K.; Dessen, P.; Couve, S.; Abdou, A.; Adam, J.; Thiery, J.; Savagner, P.; Chouaib, S. The Most Common VHL Point Mutation R167Q in Hereditary VHL Disease Interferes with Cell Plasticity Regulation. Cancers 2021, 13, 3897. [Google Scholar] [CrossRef]
- Hoffman, M.A.; Ohh, M.; Yang, H.; Klco, J.M.; Ivan, M.; Kaelin, W.G., Jr. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum. Mol. Genet. 2001, 10, 1019–1027. [Google Scholar] [CrossRef]
- Lee, S.; Nakamura, E.; Yang, H.; Wei, W.; Linggi, M.S.; Sajan, M.P.; Farese, R.V.; Freeman, R.S.; Carter, B.D.; Kaelin, W.G., Jr.; et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: Developmental culling and cancer. Cancer Cell 2005, 8, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Guo, K.; Wei, Y.; Wang, Z.; Zhang, X.; Zhang, X.; Liu, X.; Wu, W.; Wu, Z.; Zhang, L.; Cui, C.P. Deubiquitylase OTUD6B stabilizes the mutated pVHL and suppresses cell migration in clear cell renal cell carcinoma. Cell Death Dis. 2022, 13, 97. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Ma, K.; Zhou, J.; Zhang, J.; Wang, J.; Liu, S.; Zhang, Z.; Cai, L.; Zhang, N.; Gong, K. Frequent Mutations of VHL Gene and the Clinical Phenotypes in the Largest Chinese Cohort With Von Hippel-Lindau Disease. Front. Genet. 2019, 10, 867. [Google Scholar] [CrossRef]
- Miller, F.; Kentsis, A.; Osman, R.; Pan, Z.Q. Inactivation of VHL by tumorigenic mutations that disrupt dynamic coupling of the pVHL.hypoxia-inducible transcription factor-1alpha complex. J. Biol. Chem. 2005, 280, 7985–7996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, J.R.; Worth, C.L.; Bickerton, G.R.; Eisen, T.G.; Blundell, T.L. Structural bioinformatics mutation analysis reveals genotype-phenotype correlations in von Hippel-Lindau disease and suggests molecular mechanisms of tumorigenesis. Proteins 2009, 77, 84–96. [Google Scholar] [CrossRef]
- Hwang, S.; Ku, C.R.; Lee, J.I.; Hur, K.Y.; Lee, M.S.; Lee, C.H.; Koo, K.Y.; Lee, J.S.; Rhee, Y. Germline mutation of Glu70Lys is highly frequent in Korean patients with von Hippel-Lindau (VHL) disease. J. Hum. Genet. 2014, 59, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Hergovich, A.; Lisztwan, J.; Barry, R.; Ballschmieter, P.; Krek, W. Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat. Cell Biol. 2003, 5, 64–70. [Google Scholar] [CrossRef]
- Lin, G.; Zhao, Y.; Zhang, Z.; Zhang, H. Clinical diagnosis, treatment and screening of the VHL gene in three von Hippel-Lindau disease pedigrees. Exp. Ther. Med. 2020, 20, 1237–1244. [Google Scholar] [CrossRef]
- Li, Z.; Na, X.; Wang, D.; Schoen, S.R.; Messing, E.M.; Wu, G. Ubiquitination of a novel deubiquitinating enzyme requires direct binding to von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2002, 277, 4656–4662. [Google Scholar] [CrossRef] [Green Version]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaux, E.C.; Wood, S.M.; Cockman, M.E.; Nicholls, L.G.; Yeates, K.M.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J. Selection of mutant CHO cells with constitutive activation of the HIF system and inactivation of the von Hippel-Lindau tumor suppressor. J. Biol. Chem. 2001, 276, 44323–44330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knauth, K.; Bex, C.; Jemth, P.; Buchberger, A. Renal cell carcinoma risk in type 2 von Hippel-Lindau disease correlates with defects in pVHL stability and HIF-1alpha interactions. Oncogene 2006, 25, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Roberts, A.M.; Chow, J.; Coady-Osberg, N.; Ohh, M. Homotypic association between tumour-associated VHL proteins leads to the restoration of HIF pathway. Oncogene 2006, 25, 3079–3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zhang, L.; Zhang, X.; Yan, Q.; Minamishima, Y.A.; Olumi, A.F.; Mao, M.; Bartz, S.; Kaelin, W.G., Jr. Hypoxia-inducible factor linked to differential kidney cancer risk seen with type 2A and type 2B VHL mutations. Mol. Cell Biol. 2007, 27, 5381–5392. [Google Scholar] [CrossRef] [Green Version]
- Feldman, D.E.; Spiess, C.; Howard, D.E.; Frydman, J. Tumorigenic mutations in VHL disrupt folding in vivo by interfering with chaperonin binding. Mol. Cell 2003, 12, 1213–1224. [Google Scholar] [CrossRef]
- Hansen, W.J.; Ohh, M.; Moslehi, J.; Kondo, K.; Kaelin, W.G.; Welch, W.J. Diverse effects of mutations in exon II of the von Hippel-Lindau (VHL) tumor suppressor gene on the interaction of pVHL with the cytosolic chaperonin and pVHL-dependent ubiquitin ligase activity. Mol. Cell Biol. 2002, 22, 1947–1960. [Google Scholar] [CrossRef] [Green Version]
- Shmueli, M.D.; Schnaider, L.; Rosenblum, D.; Herzog, G.; Gazit, E.; Segal, D. Structural insights into the folding defects of oncogenic pVHL lead to correction of its function in vitro. PLoS ONE 2013, 8, e66333. [Google Scholar] [CrossRef] [Green Version]
- Shmueli, M.D.; Levy-Kanfo, L.; Haj, E.; Schoenfeld, A.R.; Gazit, E.; Segal, D. Arginine refolds, stabilizes, and restores function of mutant pVHL proteins in animal model of the VHL cancer syndrome. Oncogene 2019, 38, 1038–1049. [Google Scholar] [CrossRef]
- Ohh, M.; Takagi, Y.; Aso, T.; Stebbins, C.E.; Pavletich, N.P.; Zbar, B.; Conaway, R.C.; Conaway, J.W.; Kaelin, W.G., Jr. Synthetic peptides define critical contacts between elongin C, elongin B, and the von Hippel-Lindau protein. J. Clin. Investig. 1999, 104, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Thoma, C.R.; Matov, A.; Gutbrodt, K.L.; Hoerner, C.R.; Smole, Z.; Krek, W.; Danuser, G. Quantitative image analysis identifies pVHL as a key regulator of microtubule dynamic instability. J. Cell Biol. 2010, 190, 991–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couve, S.; Ladroue, C.; Laine, E.; Mahtouk, K.; Guegan, J.; Gad, S.; Le Jeune, H.; Le Gentil, M.; Nuel, G.; Kim, W.Y.; et al. Genetic evidence of a precisely tuned dysregulation in the hypoxia signaling pathway during oncogenesis. Cancer Res. 2014, 74, 6554–6564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iturrioz, X.; Parker, P.J. PKCzetaII is a target for degradation through the tumour suppressor protein pVHL. FEBS Lett. 2007, 581, 1397–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.S.; Lee, S.J.; Yoon, M.H.; Ha, N.C.; Park, B.J. Estrogen receptor α is a novel target of the Von Hippel-Lindau protein and is responsible for the proliferation of VHL-deficient cells under hypoxic conditions. Cell Cycle 2012, 11, 4462–4473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arreola, A.; Payne, L.B.; Julian, M.H.; de Cubas, A.A.; Daniels, A.B.; Taylor, S.; Zhao, H.; Darden, J.; Bautch, V.L.; Rathmell, W.K.; et al. Von Hippel-Lindau mutations disrupt vascular patterning and maturation via Notch. JCI Insight 2018, 3, e92193. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, W.K.; Hickey, M.M.; Bezman, N.A.; Chmielecki, C.A.; Carraway, N.C.; Simon, M.C. In vitro and in vivo models analyzing von Hippel-Lindau disease-specific mutations. Cancer Res. 2004, 64, 8595–8603. [Google Scholar] [CrossRef] [Green Version]
- Kurban, G.; Hudon, V.; Duplan, E.; Ohh, M.; Pause, A. Characterization of a von Hippel Lindau pathway involved in extracellular matrix remodeling, cell invasion, and angiogenesis. Cancer Res. 2006, 66, 1313–1319. [Google Scholar] [CrossRef] [Green Version]
- Tedesco, L.; Elguero, B.; Pacin, D.G.; Senin, S.; Pollak, C.; Garcia Marchinena, P.A.; Jurado, A.M.; Isola, M.; Labanca, M.J.; Palazzo, M.; et al. von Hippel-Lindau mutants in renal cell carcinoma are regulated by increased expression of RSUME. Cell Death Dis. 2019, 10, 266. [Google Scholar] [CrossRef] [Green Version]
- de Rojas, P.I.; Albinana, V.; Taranets, L.; Recio-Poveda, L.; Cuesta, A.M.; Popov, N.; Kronenberger, T.; Botella, L.M. The Endothelial Landscape and Its Role in Von Hippel-Lindau Disease. Cells 2021, 10, 2313. [Google Scholar] [CrossRef]
- Glasker, S.; Vergauwen, E.; Koch, C.A.; Kutikov, A.; Vortmeyer, A.O. Von Hippel-Lindau Disease: Current Challenges and Future Prospects. Onco Targets Ther. 2020, 13, 5669–5690. [Google Scholar] [CrossRef]
- Creighton, C.J.; Morgan, M.; Gunaratne, P.H.; Wheeler, D.A.; Gibbs, R.A.; Gordon Robertson, A.; Chu, A.; Beroukhim, R.; Cibulskis, K.; Signoretti, S.; et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Krieg, M.; Marti, H.H.; Plate, K.H. Coexpression of erythropoietin and vascular endothelial growth factor in nervous system tumors associated with von Hippel-Lindau tumor suppressor gene loss of function. Blood 1998, 92, 3388–3393. [Google Scholar] [CrossRef] [PubMed]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R.; Prchal, J.T. Vascular complications in Chuvash polycythemia. Semin. Thromb. Hemost. 2006, 32, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem. Biophys. Res. Commun. 2005, 338, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.H.; Knudson, A.G.; Pandolfi, P.P. A continuum model for tumour suppression. Nature 2011, 476, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Ang, S.O.; Chen, H.; Gordeuk, V.R.; Sergueeva, A.I.; Polyakova, L.A.; Miasnikova, G.Y.; Kralovics, R.; Stockton, D.W.; Prchal, J.T. Endemic polycythemia in Russia: Mutation in the VHL gene. Blood Cells Mol. Dis. 2002, 28, 57–62. [Google Scholar] [CrossRef]
- Polyakova, L.A. Familial erythrocytosis among inhabitants of the Chuvash ASSR. Probl. Gematol. Pereliv. Krovi 1974, 10, 30–36. [Google Scholar]
- Sergeyeva, A.; Gordeuk, V.R.; Tokarev, Y.N.; Sokol, L.; Prchal, J.F.; Prchal, J.T. Congenital polycythemia in Chuvashia. Blood 1997, 89, 2148–2154. [Google Scholar] [CrossRef] [Green Version]
- Maher, E.R.; Neumann, H.P.; Richard, S. von Hippel-Lindau disease: A clinical and scientific review. Eur. J. Hum. Genet. 2011, 19, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Klingler, J.H.; Glasker, S.; Bausch, B.; Urbach, H.; Krauss, T.; Jilg, C.A.; Steiert, C.; Puzik, A.; Neumann-Haefelin, E.; Kotsis, F.; et al. Hemangioblastoma and von Hippel-Lindau disease: Genetic background, spectrum of disease, and neurosurgical treatment. Childs Nerv. Syst. 2020, 36, 2537–2552. [Google Scholar] [CrossRef] [PubMed]
- Catapano, D.; Muscarella, L.A.; Guarnieri, V.; Zelante, L.; D’Angelo, V.A.; D’Agruma, L. Hemangioblastomas of central nervous system: Molecular genetic analysis and clinical management. Neurosurgery 2005, 56, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Ma, K.; Zhang, J.; Hong, B.; Zhou, J.; Li, L.; Zhang, K.; Gong, K.; Cai, L. Novel genetic characterisation and phenotype correlation in von Hippel-Lindau (VHL) disease based on the Elongin C binding site: A large retrospective study. J. Med. Genet. 2020, 57, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Karimi, S.; Arabi, A.; Shahraki, T.; Safi, S. Von Hippel-Lindau Disease and the Eye. J. Ophthalmic Vis. Res. 2020, 15, 78. [Google Scholar] [CrossRef]
- Ruppert, M.D.; Gavin, M.; Mitchell, K.T.; Peiris, A.N. Ocular Manifestations of von Hippel-Lindau Disease. Cureus 2019, 11, e5319. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.D.; Shields, C.L.; Shields, J.A. von Hippel-Lindau disease. Surv. Ophthalmol. 2001, 46, 117–142. [Google Scholar] [CrossRef]
- Chew, E.Y. Ocular manifestations of von Hippel-Lindau disease: Clinical and genetic investigations. Trans. Am. Ophthalmol. Soc. 2005, 103, 495–511. [Google Scholar]
- Hajjaj, A.; van Overdam, K.A.; Oldenburg, R.A.; Koopmans, A.E.; van den Ouweland, A.M.W.; de Klein, A.; Kilic, E. Retinal haemangioblastomas in von Hippel-Lindau germline mutation carriers: Progression, complications and treatment outcome. Acta Ophthalmol. 2020, 98, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.T.; Agron, E.; Coleman, H.R.; Reed, G.F.; Csaky, K.; Peterson, J.; Glenn, G.; Linehan, W.M.; Albert, P.; Chew, E.Y. Genotype-phenotype correlation in von Hippel-Lindau disease with retinal angiomatosis. Arch. Ophthalmol. 2007, 125, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.T.; Agron, E.; Coleman, H.R.; Tran, T.; Reed, G.F.; Csaky, K.; Chew, E.Y. Clinical characterization of retinal capillary hemangioblastomas in a large population of patients with von Hippel-Lindau disease. Ophthalmology 2008, 115, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Toy, B.C.; Agron, E.; Nigam, D.; Chew, E.Y.; Wong, W.T. Longitudinal analysis of retinal hemangioblastomatosis and visual function in ocular von Hippel-Lindau disease. Ophthalmology 2012, 119, 2622–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, D.; Neumann, H.P. Retinal vascular hamartoma in von Hippel-Lindau disease. Arch. Ophthalmol. 1995, 113, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- de Jong, P.T.; Verkaart, R.J.; van de Vooren, M.J.; Majoor-Krakauer, D.F.; Wiegel, A.R. Twin vessels in von Hippel-Lindau disease. Am. J. Ophthalmol. 1988, 105, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.T.; Yeh, S.; Chan, C.C.; Kalina, R.E.; Kinyoun, J.L.; Folk, J.C.; Coleman, H.R.; Chew, E.Y. Retinal vascular proliferation as an ocular manifestation of von Hippel-Lindau disease. Arch. Ophthalmol. 2008, 126, 637–643. [Google Scholar] [CrossRef] [Green Version]
- Wittstrom, E.; Nordling, M.; Andreasson, S. Genotype-phenotype correlations, and retinal function and structure in von Hippel-Lindau disease. Ophthalm. Genet. 2014, 35, 91–106. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, J.C.; Zeng, R.; Nagata, T.; Katz, R.; Mukai, S.; Miller, J.B. Detection of retinal microvascular changes in von Hippel-Lindau disease using optical coherence tomography angiography. PLoS ONE 2020, 15, e0229213. [Google Scholar] [CrossRef] [Green Version]
- Pilotto, E.; Nacci, E.B.; Ferrara, A.M.; De Moja, G.; Zovato, S.; Midena, E. Macular Perfusion Impairment in Von Hippel-Lindau Disease Suggests a Generalized Retinal Vessel Alteration. J. Clin. Med. 2020, 9, 2677. [Google Scholar] [CrossRef]
- Pilotto, E.; Nacci, E.B.; De Moja, G.; Ferrara, A.M.; Parrozzani, R.; Londei, D.; Zovato, S.; Midena, E. Structural and microvascular changes of the peripapillary retinal nerve fiber layer in Von Hippel-Lindau disease: An OCT and OCT angiography study. Sci. Rep. 2021, 11, 25. [Google Scholar] [CrossRef]
- Boratto, S.D.F.; Cardoso, P.A.S.; Priolli, D.G.; Botelho, R.V.; Goldenberg, A.; Bianco, B.; Waisberg, J. von Hippel-Lindau Syndrome: Genetic Study of Case With a Rare Pathogenic Variant With Optic Nerve Hemangioblastoma, a Rare Phenotypic Expression. Front. Oncol. 2020, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Li, Z.; Wang, Y.; Zhang, C.; Zhang, Z.; Zhang, X. Central Nervous System Hemangioblastoma in a Pediatric Patient Associated With Von Hippel-Lindau Disease: A Case Report and Literature Review. Front. Oncol. 2021, 11, 683021. [Google Scholar] [CrossRef]
- Chen, S.; Chew, E.Y.; Chan, C.C. Pathology characteristics of ocular von Hippel-Lindau disease with neovascularization of the iris and cornea: A case report. J. Med. Case Rep. 2015, 9, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dollfus, H.; Massin, P.; Taupin, P.; Nemeth, C.; Amara, S.; Giraud, S.; Beroud, C.; Dureau, P.; Gaudric, A.; Landais, P.; et al. Retinal hemangioblastoma in von Hippel-Lindau disease: A clinical and molecular study. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3067–3074. [Google Scholar]
- Mettu, P.; Agron, E.; Samtani, S.; Chew, E.Y.; Wong, W.T. Genotype-phenotype correlation in ocular von Hippel-Lindau (VHL) disease: The effect of missense mutation position on ocular VHL phenotype. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4464–4470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscheck, F.; Fraune, C.; Simon, R.; Kluth, M.; Hube-Magg, C.; Moller-Koop, C.; Sarper, I.; Ketterer, K.; Henke, T.; Eichelberg, C.; et al. Prevalence and clinical significance of VHL mutations and 3p25 deletions in renal tumor subtypes. Oncotarget 2020, 11, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, A.A.; Nakamura, E.; Qi, J.; Creech, A.; Jaffe, J.D.; Paulk, J.; Novak, J.S.; Nagulapalli, K.; McBrayer, S.K.; Cowley, G.S.; et al. HIF activation causes synthetic lethality between the VHL tumor suppressor and the EZH1 histone methyltransferase. Sci. Transl. Med. 2017, 9, eaal5272. [Google Scholar] [CrossRef] [Green Version]
- Jonasch, E.; Walker, C.L.; Rathmell, W.K. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat. Rev. Nephrol. 2021, 17, 245–261. [Google Scholar] [CrossRef]
- Gao, W.; Li, W.; Xiao, T.; Liu, X.S.; Kaelin, W.G., Jr. Inactivation of the PBRM1 tumor suppressor gene amplifies the HIF-response in VHL-/- clear cell renal carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Testa, U.; Pelosi, E.; Castelli, G. Genetic Alterations in Renal Cancers: Identification of The Mechanisms Underlying Cancer Initiation and Progression and of Therapeutic Targets. Medicines 2020, 7, 44. [Google Scholar] [CrossRef]
- Gerlinger, M.; Horswell, S.; Larkin, J.; Rowan, A.J.; Salm, M.P.; Varela, I.; Fisher, R.; McGranahan, N.; Matthews, N.; Santos, C.R.; et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014, 46, 225–233. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef]
- Ayala-Ramirez, M.; Feng, L.; Johnson, M.M.; Ejaz, S.; Habra, M.A.; Rich, T.; Busaidy, N.; Cote, G.J.; Perrier, N.; Phan, A.; et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: Primary tumor size and primary tumor location as prognostic indicators. J. Clin. Endocrinol. Metab. 2011, 96, 717–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, S.; Zhang, J.; Tan, X.; Huang, Y.; Xu, J.; Silk, N.; Zhang, D.; Liu, Q.; Jiang, J. The VHL/HIF Axis in the Development and Treatment of Pheochromocytoma/Paraganglioma. Front. Endocrinol. 2020, 11, 586857. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Carbonero, R.; Matute Teresa, F.; Mercader-Cidoncha, E.; Mitjavila-Casanovas, M.; Robledo, M.; Tena, I.; Alvarez-Escola, C.; Aristegui, M.; Bella-Cueto, M.R.; Ferrer-Albiach, C.; et al. Multidisciplinary practice guidelines for the diagnosis, genetic counseling and treatment of pheochromocytomas and paragangliomas. Clin. Transl. Oncol. 2021, 23, 1995–2019. [Google Scholar] [CrossRef] [PubMed]
- Darr, R.; Kater, J.; Sekula, P.; Bausch, B.; Krauss, T.; Bode, C.; Walz, G.; Neumann, H.P.; Zschiedrich, S. Clinical decision making in small non-functioning VHL-related incidentalomas. Endocr. Connect. 2020, 9, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Guha, A.; Vicha, A.; Zelinka, T.; Musil, Z.; Chovanec, M. Genetic Variants in Patients with Multiple Head and Neck Paragangliomas: Dilemma in Management. Biomedicines 2021, 9, 626. [Google Scholar] [CrossRef]
- Gimenez-Roqueplo, A.P.; Dahia, P.L.; Robledo, M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm. Metab. Res. 2012, 44, 328–333. [Google Scholar] [CrossRef]
- Lopez-Jimenez, E.; Gomez-Lopez, G.; Leandro-Garcia, L.J.; Munoz, I.; Schiavi, F.; Montero-Conde, C.; de Cubas, A.A.; Ramires, R.; Landa, I.; Leskela, S.; et al. Research resource: Transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol. Endocrinol. 2010, 24, 2382–2391. [Google Scholar] [CrossRef] [Green Version]
- Tsoli, M.; Daskalakis, K.; Kassi, E.; Kaltsas, G.; Tsolakis, A.V. A Critical Appraisal of Contemporary and Novel Biomarkers in Pheochromocytomas and Adrenocortical Tumors. Biology 2021, 10, 580. [Google Scholar] [CrossRef]
- Gao, S.; Liu, L.; Li, Z.; Pang, Y.; Shi, J.; Zhu, F. Seven Novel Genes Related to Cell Proliferation and Migration of VHL-Mutated Pheochromocytoma. Front. Endocrinol. 2021, 12, 598656. [Google Scholar] [CrossRef]
- Dong, Y.; Huang, Y.; Fan, C.; Wang, L.; Zhang, R.; Li, W.; Guo, Z.; Wang, D.; Zheng, Z. GIPC2 is an endocrine-specific tumor suppressor gene for both sporadic and hereditary tumors of RET- and SDHB-, but not VHL-associated clusters of pheochromocytoma/paraganglioma. Cell Death Dis. 2021, 12, 444. [Google Scholar] [CrossRef]
- Guha, A.; Musil, Z.; Vicha, A.; Zelinka, T.; Pacak, K.; Astl, J.; Chovanec, M. A systematic review on the genetic analysis of paragangliomas: Primarily focused on head and neck paragangliomas. Neoplasma 2019, 66, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Castro-Teles, J.; Sousa-Pinto, B.; Rebelo, S.; Pignatelli, D. Pheochromocytomas and paragangliomas in von Hippel-Lindau disease: Not a needle in a haystack. Endocr. Connect. 2021, 10, R293–R304. [Google Scholar] [CrossRef] [PubMed]
- Flores, S.K.; Cheng, Z.; Jasper, A.M.; Natori, K.; Okamoto, T.; Tanabe, A.; Gotoh, K.; Shibata, H.; Sakurai, A.; Nakai, T.; et al. A synonymous VHL variant in exon 2 confers susceptibility to familial pheochromocytoma and von Hippel-Lindau disease. J. Clin. Endocrinol. Metab. 2019, 104, 3826–3834. [Google Scholar] [CrossRef]
- Buffet, A.; Calsina, B.; Flores, S.; Giraud, S.; Lenglet, M.; Romanet, P.; Deflorenne, E.; Aller, J.; Bourdeau, I.; Bressac-de Paillerets, B.; et al. Germline mutations in the new E1′ cryptic exon of the VHL gene in patients with tumours of von Hippel-Lindau disease spectrum or with paraganglioma. J. Med. Genet. 2020, 57, 752–759. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, S.; Mukasa, A.; Tanaka, S.; Nomura, M.; Omata, M.; Yanagisawa, S.; Yamamoto, S.; Ichimura, K.; Nakatomi, H.; Ueki, K.; et al. Differences in genetic and epigenetic alterations between von Hippel-Lindau disease-related and sporadic hemangioblastomas of the central nervous system. Neuro. Oncol. 2017, 19, 1228–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olschwang, S.; Richard, S.; Boisson, C.; Giraud, S.; Laurent-Puig, P.; Resche, F.; Thomas, G. Germline mutation profile of the VHL gene in von Hippel-Lindau disease and in sporadic hemangioblastoma. Hum. Mutat. 1998, 12, 424–430. [Google Scholar] [CrossRef]
- Stolle, C.; Glenn, G.; Zbar, B.; Humphrey, J.S.; Choyke, P.; Walther, M.; Pack, S.; Hurley, K.; Andrey, C.; Klausner, R.; et al. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum. Mutat. 1998, 12, 417–423. [Google Scholar] [CrossRef]
- McMullin, M.F. The classification and diagnosis of erythrocytosis. Int. J. Lab. Hematol. 2008, 30, 447–459. [Google Scholar] [CrossRef]
- Wouters, H.; Mulder, R.; van Zeventer, I.A.; Schuringa, J.J.; van der Klauw, M.M.; van der Harst, P.; Diepstra, A.; Mulder, A.B.; Huls, G. Erythrocytosis in the general population: Clinical characteristics and association with clonal hematopoiesis. Blood Adv. 2020, 4, 6353–6363. [Google Scholar] [CrossRef]
- Nguyen, E.; Harnois, M.; Busque, L.; Sirhan, S.; Assouline, S.; Chamaki, I.; Olney, H.; Mollica, L.; Szuber, N. Phenotypical differences and thrombosis rates in secondary erythrocytosis versus polycythemia vera. Blood Cancer J. 2021, 11, 75. [Google Scholar] [CrossRef]
- Ascher, K.W. Eye manifestations in polycythemia. JAMA 1971, 215, 295. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Neriyanuri, S.; Raman, R. Management of macular edema with branch retinal vein occlusion in a case of secondary polycythemia. GMS Ophthalmol. Cases 2019, 9, Doc38. [Google Scholar] [CrossRef] [PubMed]
- Liisborg, C.; Hasselbalch, H.C.; Sorensen, T.L. Ocular Manifestations in Patients with Philadelphia-Negative Myeloproliferative Neoplasms. Cancers 2020, 12, 573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.S.; Joe, S.G.; Kim, J.G.; Park, S.H.; Ko, H.S. Delayed choroidal and retinal blood flow in polycythaemia vera patients with transient ocular blindness: A preliminary study with fluorescein angiography. Br. J. Haematol. 2013, 161, 745–747. [Google Scholar] [CrossRef] [PubMed]
- McMullin, M.F. Genetic Background of Congenital Erythrocytosis. Genes 2021, 12, 1151. [Google Scholar] [CrossRef] [PubMed]
- Pastore, Y.; Jedlickova, K.; Guan, Y.; Liu, E.; Fahner, J.; Hasle, H.; Prchal, J.F.; Prchal, J.T. Mutations of von Hippel-Lindau tumor-suppressor gene and congenital polycythemia. Am. J. Hum. Genet. 2003, 73, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.J.; McMullin, M.F.; Jowitt, S.N.; Potter, M.; Treacy, M.; Watson, W.H.; Lappin, T.R. Chuvash-type congenital polycythemia in 4 families of Asian and Western European ancestry. Blood 2003, 102, 1097–1099. [Google Scholar] [CrossRef] [Green Version]
- Gordeuk, V.R.; Sergueeva, A.I.; Miasnikova, G.Y.; Okhotin, D.; Voloshin, Y.; Choyke, P.L.; Butman, J.A.; Jedlickova, K.; Prchal, J.T.; Polyakova, L.A. Congenital disorder of oxygen sensing: Association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood 2004, 103, 3924–3932. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The VHL Tumor Suppressor Gene: Insights into Oxygen Sensing and Cancer. Trans. Am. Clin. Climatol. Assoc. 2017, 128, 298–307. [Google Scholar]
- Tomasic, N.L.; Piterkova, L.; Huff, C.; Bilic, E.; Yoon, D.; Miasnikova, G.Y.; Sergueeva, A.I.; Niu, X.; Nekhai, S.; Gordeuk, V.; et al. The phenotype of polycythemia due to Croatian homozygous VHL (571C>G:H191D) mutation is different from that of Chuvash polycythemia (VHL 598C>T:R200W). Haematologica 2013, 98, 560–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapralova, K.; Lanikova, L.; Lorenzo, F.; Song, J.; Horvathova, M.; Divoky, V.; Prchal, J.T. RUNX1 and NF-E2 upregulation is not specific for MPNs, but is seen in polycythemic disorders with augmented HIF signaling. Blood 2014, 123, 391–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarangi, S.; Lanikova, L.; Kapralova, K.; Acharya, S.; Swierczek, S.; Lipton, J.M.; Wolfe, L.; Prchal, J.T. The homozygous VHL(D126N) missense mutation is associated with dramatically elevated erythropoietin levels, consequent polycythemia, and early onset severe pulmonary hypertension. Pediatr. Blood Cancer 2014, 61, 2104–2106. [Google Scholar] [CrossRef]
- Lanikova, L.; Lorenzo, F.; Yang, C.; Vankayalapati, H.; Drachtman, R.; Divoky, V.; Prchal, J.T. Novel homozygous VHL mutation in exon 2 is associated with congenital polycythemia but not with cancer. Blood 2013, 121, 3918–3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remenyi, G.; Bereczky, Z.; Gindele, R.; Ujfalusi, A.; Illes, A.; Udvardy, M. rs779805 Von Hippel-Lindau Gene Polymorphism Induced/Related Polycythemia Entity, Clinical Features, Cancer Association, and Familiar Characteristics. Pathol. Oncol. Res. 2021, 27, 1609987. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of VHL gene structure. (A). VHL gene structure and protein isoforms with corresponding VHL transcripts. (B). Putative VHL transcripts, containing exon E′, identified by cloning and sequencing [15].
Figure 1.
Schematic representation of VHL gene structure. (A). VHL gene structure and protein isoforms with corresponding VHL transcripts. (B). Putative VHL transcripts, containing exon E′, identified by cloning and sequencing [15].

Figure 2.
Role of VHL in adaptive response to oxygen levels.

Figure 3.
STRING network of VHL protein–protein interactions. Settings: Network type, full STRING network; meaning of network edges, confidence (line thickness indicates the strength of data support); active interaction sources, experiments; minimum required interaction score, high confidence (0.700); max number of interactors to show, no more than 20 interactors; organism, Homo sapiens [78].
Figure 3.
STRING network of VHL protein–protein interactions. Settings: Network type, full STRING network; meaning of network edges, confidence (line thickness indicates the strength of data support); active interaction sources, experiments; minimum required interaction score, high confidence (0.700); max number of interactors to show, no more than 20 interactors; organism, Homo sapiens [78].

Figure 4.
MRI imaging showing CNS lesions of the patient consistent with hemangioblastomas (yellow arrows): (A) Lesion pressing the brainstem and upper spinal cord; (B) Cerebellar lesion; (C) Spinal cord lesion.
Figure 4.
MRI imaging showing CNS lesions of the patient consistent with hemangioblastomas (yellow arrows): (A) Lesion pressing the brainstem and upper spinal cord; (B) Cerebellar lesion; (C) Spinal cord lesion.

Figure 5.
Fluorescein angiography image of the patient’s left eye fundus; yellow arrows show retinal hemangioblastomas.
Figure 5.
Fluorescein angiography image of the patient’s left eye fundus; yellow arrows show retinal hemangioblastomas.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
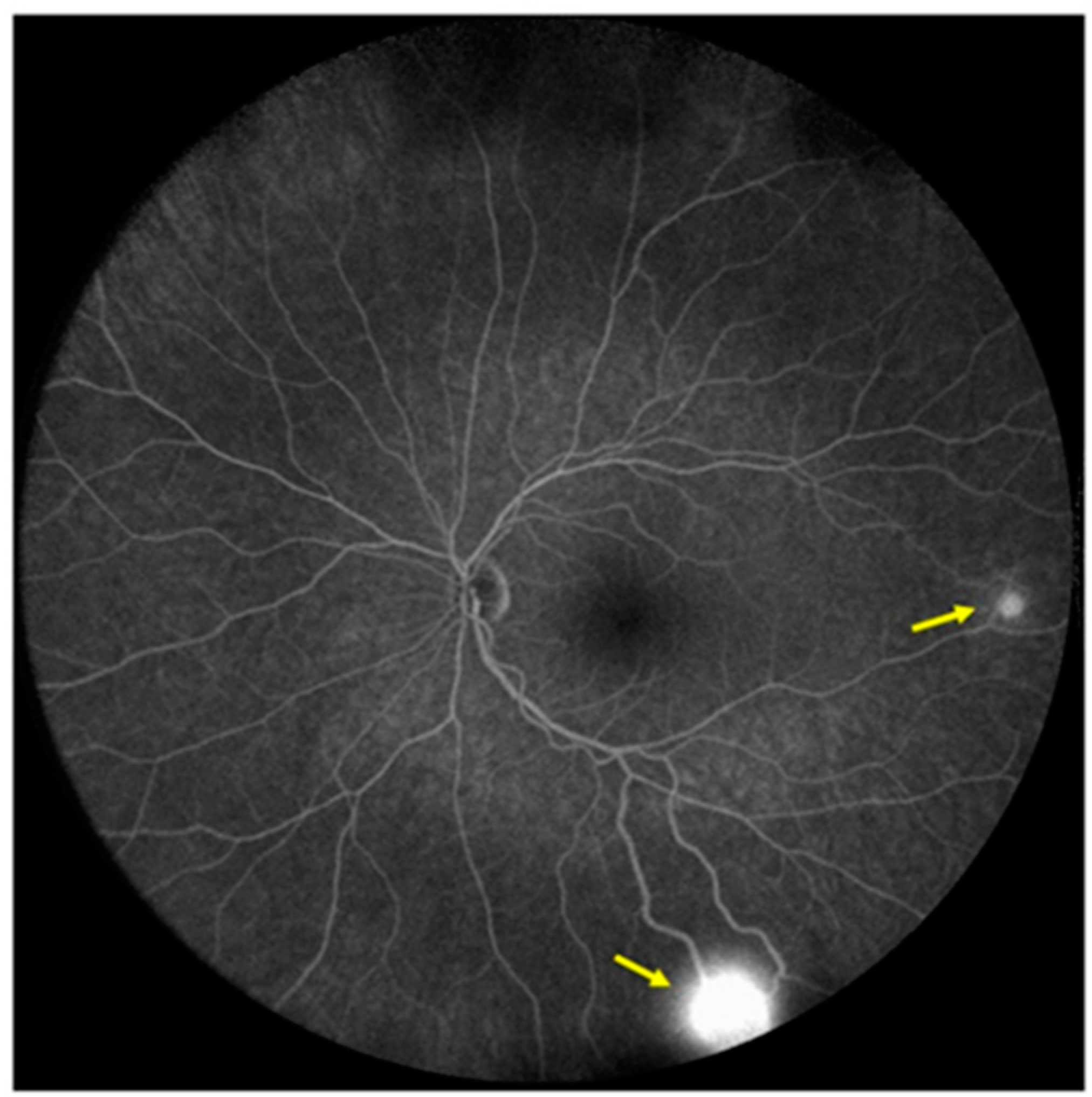{kind=link}
Table 1.
Selected VHL genetic variants in VHL disease and their association with phenotype.
| Variant | Protein Change | Codon | VHL Type/Phenotype | Functional Consequence | Reference |
|---|---|---|---|---|---|
| c.191G>C | R64P | 64 | Type 2C | Increased aPKC JUNB levels; impaired binding to fibronectin. | [91,92] |
| c.194C>T | S65L | 56 | Type 2B | Impaired HIF1α binding; impaired HIF2α regulation. | [93,94,95] |
| c.208G>A | E70K | 70 | Type 1 | Impaired HIF1α binding. | [96,97] |
| c.233A>G | N78S | 78 | Type 1 | Impaired HIF1α regulation. | [98,99] |
| c.239G>A | S80N | 80 | Type 2C | No known consequence. | [98] |
| c.245G>C | R82P | 82 | Type 2B | Loss of function of VHL. | [100] |
| c.250G>C | V84L | 84 | Type 2C | No known consequence. | [98] |
| c.262T>A c.262T>C | W88R | 88 | Hemangio- Blastoma 1 | No known consequence. | [94] |
| c.269A>T | N90I | 90 | Type 2B | Impaired HIF1α regulation. | [94,98,101,102] |
| c.292T>C | Y98H | 98 | Type 2A | Impaired HIF1α regulation; defective microtubule stabilization. | [91,98] |
| c.292T>A | Y98N | 98 | Type 2B | Impaired HIF1α regulation; impaired GLUT1 suppression. | [101] |
| c.334T>A | Y112H | 112 | Type 2A | Impaired HIF1α regulation; decreased VHL stability. | [89,103] |
| c.334T>A | Y112N | 112 | Type 2B | Reduced stability of the Vhl-Elongin B/C complex; impaired HIF1α regulation; elevated HIF2α, GLUT1, and cyclin D1 expression in normoxic conditions. | [103,104,105] |
| c.334T>G | Y112D | 112 | Type 2C | No known consequence. | [98] |
| c.340G>C | G114R | 114 | Type 2B | Reduced stability of the Vhl-Elongin B/C complex. | [106] |
| c.349T>C c.349T>A | W117R | 117 | Type 2B | Impaired HIF1α regulation; impaired binding to fibronectin; elevated HIF2α and GLUT1 expression in normoxic conditions. | [62,105,107] |
| c.355T>C c.357C>G c.357C>A | F119L | 119 | Type 2B | Decreased VHL stability; impaired HIF1α regulation. | [108] |
| c.407T>C | F136S | 136 | Type 2B | No known consequence. | [98] |
| c.407T>A | F136Y | 136 | Type 2B | No known consequence. | [98] |
| c.408T>G | F136L | 136 | Type 2B | Decreased VHL stability; impaired HIF1α regulation. | [108,109] |
| c.482G>C | R161P | 161 | Type 2B | Reduced stability of the Vhl-Elongin B/C complex; defective microtubule stabilization. | [110,111] |
| c.482G>A | R161Q | 161 | Type 2A; Type 2B | Reduced VHL stability. | [112] |
| c.486C>G | C162W | 162 | Hemangio- Blastoma 1 | Impaired HIF1α regulation. | [94,113] |
| c.499C>T | R167W | 167 | Type 2B | Decreased binding to Elongin B/C and Cullin-2; impaired ubiquitination and degradation of ESR1. | [62,114] |
| c.500G>A | R167Q | 167 | Hemangio- Blastoma 1 | Decreased binding to Elongin C; impaired HIF2α regulation. | [94,115,116] |
| c.562C>G | L188V | Type 2C | Impaired binding to fibronectin; elevated RWWD3, aPKC, and JUNB levels. | [91,92,117,118] |
1 type of VHL disease not classified.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hudler, P.; Urbancic, M. The Role of VHL in the Development of von Hippel-Lindau Disease and Erythrocytosis. Genes 2022, 13, 362. https://doi.org/10.3390/genes13020362
AMA Style
Hudler P, Urbancic M. The Role of VHL in the Development of von Hippel-Lindau Disease and Erythrocytosis. Genes. 2022; 13(2):362. https://doi.org/10.3390/genes13020362
Chicago/Turabian StyleHudler, Petra, and Mojca Urbancic. 2022. "The Role of VHL in the Development of von Hippel-Lindau Disease and Erythrocytosis" Genes 13, no. 2: 362. https://doi.org/10.3390/genes13020362
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.