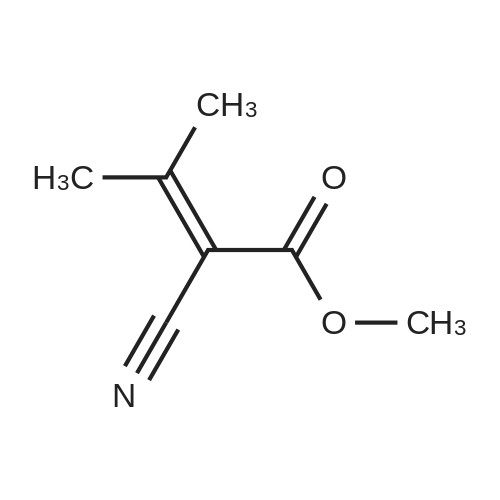
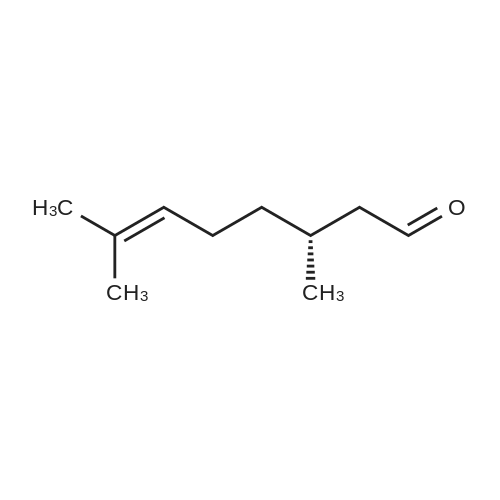
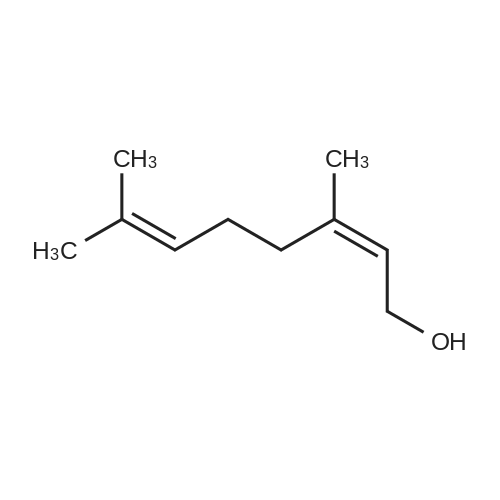
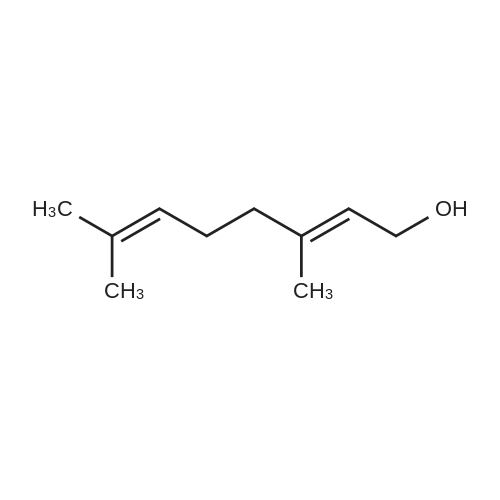
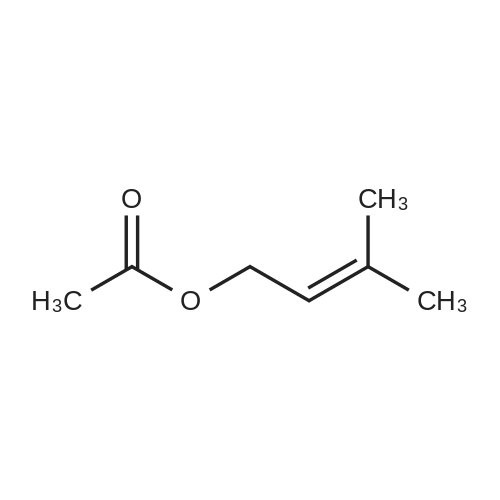
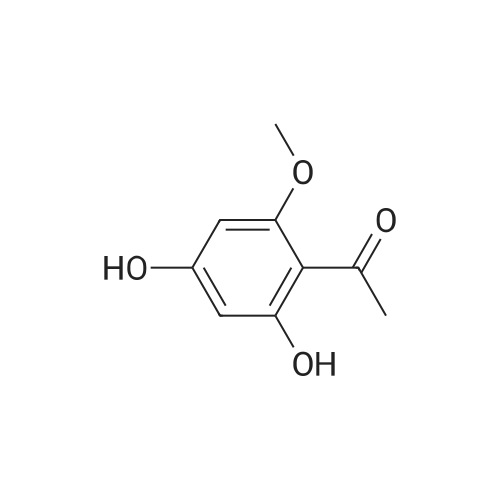
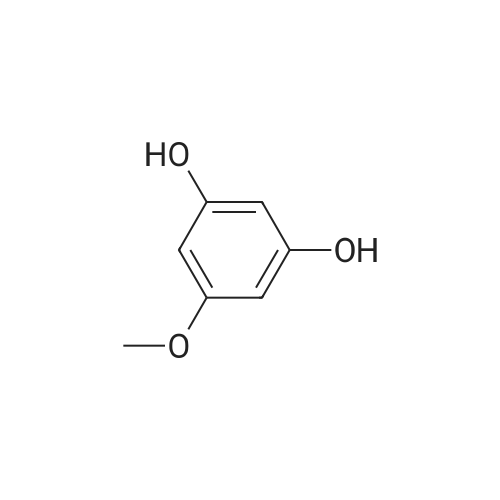
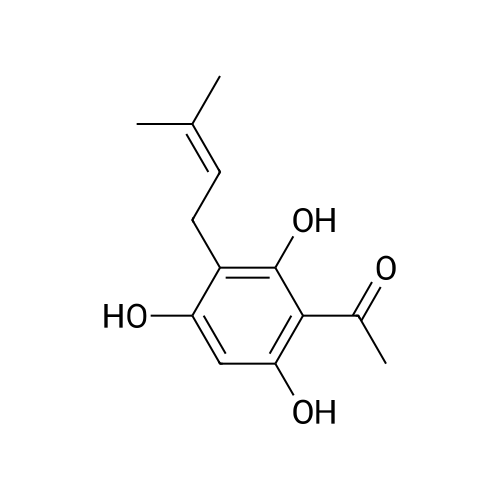
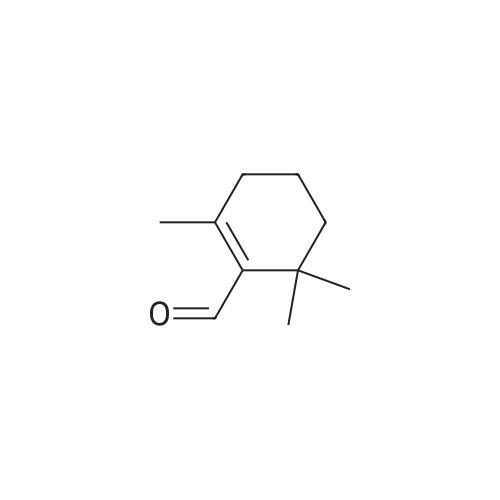
| 98% |
With C36H103AlO4Si14; isopropanol In neat (no solvent) at 50℃; for 24h; Glovebox; Schlenk technique; |
|
| 98% |
With C55H44O2P4Ru; hydrogen In toluene at 80℃; for 18h; Glovebox; Autoclave; |
4 Example 4
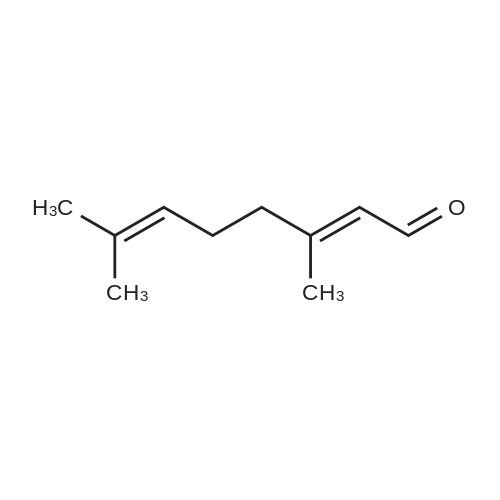
In an argon glove box,A 5 mL vial equipped with a magnetic stir bar was charged with the desired amount of catalyst 2 (0.005 mol%),Citral 3b (5 mmol) and toluene (1.5 mL) were added and the mixture was transferred to a non-polluting autoclave,Then use the H2 (10atm) pressure / exhaust three cycles for ventilation.It was then pressurized with H2 (50 atm) and disconnected from the H2 source, the autoclave was placed in an oil bath preheated to 80 ° C,After the reaction was stirred for 18h, the autoclave was cooled in an ice bath and the hydrogen was slowly released.The silica gel column gave product 4b in a yield of 98%The selectivity of the double bond without being reduced is 97%. |
| 98% |
With C55H44O2P4Ru; hydrogen In toluene at 80℃; for 18h; Inert atmosphere; Glovebox; Autoclave; |
|
| 98.85% |
With zirconium hydroxycarbonate In isopropanol at 80℃; for 15h; Autoclave; Sealed tube; |
|
| 97% |
With mesoporous silica; sodium cyanotrihydridoborate at 70 - 80℃; for 0.0138889h; Neat (no solvent); regioselective reaction; |
|
| 97% |
With sodium tetrahydridoborate; diammonium oxalate In acetonitrile at 20℃; for 0.333333h; regioselective reaction; |
|
| 97% |
With iron(II) fluoro{tris[2-(diphenylphosphino)phenyl]phospino}tetrafluoroborate; hydrogen; trifluoroacetic acid In isopropanol at 120℃; Inert atmosphere; Autoclave; chemoselective reaction; |
|
| 97% |
With C29H34BNOP2Ru In ethanol; dichloromethane Schlenk technique; Inert atmosphere; Reflux; |
|
| 97% |
With C46H49CoN3P4(2+)*2BF4(1-); hydrogen; potassium hydroxide In ethanol; acetonitrile at 60℃; for 24h; Autoclave; Glovebox; chemoselective reaction; |
|
| 96% |
With iron(II) fluoride; water monomer; aluminium for 24h; |
|
| 96% |
With di-μ-chlorobis-[(η6-p-cymene)chlororuthenium(II)]; C13H19N4O2S(1+)*ClH*Cl(1-); sodium formate dihydrate In water monomer at 80℃; for 3.5h; Inert atmosphere; chemoselective reaction; |
|
| 96% |
With hydrogen; silver(0) In tetrahydrofuran at 150℃; for 72h; |
11
Catalysts E to G for Examples obtained in Production Example 4 were each 0.008 mmol in terms of metal equivalent of silver, incidentally, as a substrate having a carbon-carbon double bond and an aldehyde group, 0.25 mmol of citral (a mixture of cis-trans isomers geranial and neral 2: 1) Solvent 5 ml of tetrahydrofuran (THF), hydrogen pressure 1.5 MPa, Hydrogenation reduction treatment was carried out at a reaction temperature of 150 ° C. reaction time is In each of Examples 11 to 13, it was 72 hours, by using gas chromatograph, yield, selectivity was measured. The results are shown in Table 5. |
| 95% |
With (Ppyz)Zr(BH4)2Cl2 In diethyl ether at 20℃; for 2h; |
|
| 95% |
Stage #1: (E/Z)-3,7-dimethyl-2,6-octadienal With [(N,N'-bis(diisopropylphosphino)-2,6-diaminopyridine)Mn(CO)2H] In 1,2-dimethoxyethane at 110℃; for 18h; Inert atmosphere; Sealed tube;
Stage #2: With sodium hydroxide at 25℃; for 18h; Inert atmosphere; chemoselective reaction; |
General procedure for hydrosilylation reactions
General procedure: Inside an Ar-flushed glovebox, an 8 cm3 microwave vial was charged with complex (0.01-0.03 mol%), carbonyl substrate (0.35 mmol), 2 cm3 solvent, and silane (0.035-0.1 mmol) in this order. A stirring bar was added, and the vial was sealed. The closed vial was removed from the glovebox and stirred for 18 h at the indicated temperature in a heated aluminum block. The vial was allowed to reach room temperature and the reaction was quenched by exposure to air. In case of screening reactions, fluorobenzene (0.35 mmol) was added and the reaction mixture was analyzed by 19F{1H} NMR. Isolation of the product To the reaction mixture 2 cm3 of a 20 wt% NaOH-solution were added and the solution was stirred for 18 h at room temperature. The phases were separated, and the aqueous phase was three times extracted with 2 cm3 diethyl ether. The combined organic phases were filtrated over a pad of silica, dried over Na2SO4 and the solvent was removed. Spectroscopic data of all isolated products are in line with the literature [11, 42-49]. |
| 94% |
With hydrogen In tetrahydrofuran at 150℃; for 12h; Autoclave; chemoselective reaction; |
|
| 94% |
With hydrogen; Cs2CO3 In tetrahydrofuran at 150℃; for 6h; Autoclave; chemoselective reaction; |
|
| 94% |
With sodium tetrahydridoborate In tetrahydrofuran at 20℃; for 0.333333h; |
|
| 94% |
With Cp*Ir(6,6'-dionato-2,2'-bipyridine)(H2O); isopropanol at 82℃; for 6h; Inert atmosphere; Green chemistry; |
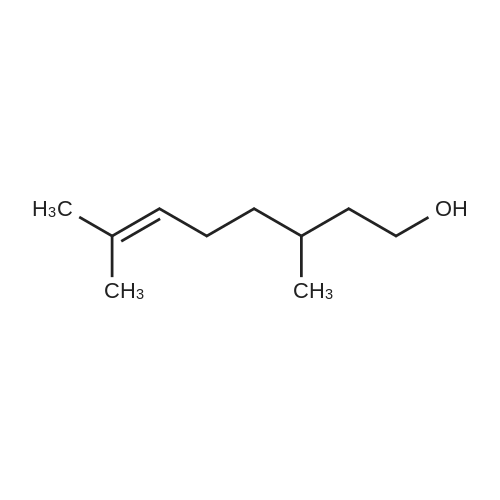
15 3,7-dimethyl-2,6-octadienol
Citral (152 mg, 1.0 mmol), cat. [Ir] (1.1 mg, 0.002 mmol, 0.2 mol%) and isopropanol (5 mL) were sequentially added to a 25 mL Kelvin tube, protected with N2 and reacted at 82 ° C for 6 h. Cool to room temperature and remove the solvent by rotary evaporation.The pure target compound was obtained by column chromatography (developing solvent: petroleum ether / ethyl acetate), yield: 94% |
| 93% |
With sodium tetrahydridoborate; poly[N-(2-aminoethyl)acrylamido]trimethyl ammonium chloride In tetrahydrofuran; water monomer at 20℃; for 0.416667h; |
|
| 93% |
With Zn(2+)*2BH4(1-)*C6H7NO In acetonitrile at 20℃; for 0.0166667h; chemoselective reaction; |
Reduction of Benzaldehyde to Benzyl alcoholwith [Zn(BH4)2(2-MeOpy)]
General procedure: A Typical ProcedureIn a round-bottomed flask (10 mL),equipped with a magnetic stirrer, a solution ofbanzaldehye (0.106 g, l mmol) in CH3CN (3 mL)was prepared. The complex reducing agent (0.1 g,0.5 mmol) was then added and the mixture wasstirred at room temperature. TLC monitored theprogress of the reaction (eluent; Hexane/EtOAc: 9/1). After completion of the reaction within 1 min, asolution of 5% HCl (5 mL) was added to the reactionmixture and stirred for 5 min. The mixture was extracted with CH2Cl2 (3 × 10 mL) and dried overthe anhydrous sodium sulfate. Evaporation of thesolvent and short column chromatography of theresulting crude material over silica gel (0.015-0.040mm) by eluent of (Hexane/EtOAc: 9/1) afforded thepure liquid benzyl alcohol (0.105 g, 98% yield) |
| 93% |
With NaNO3; sodium tetrahydridoborate; acetophenone In water monomer at 20℃; for 1h; regioselective reaction; |
|
| 92% |
With sodium tris(acetoxy)borohydride In benzene for 3h; Heating; |
|
| 92% |
With aluminum(III) oxide; zinc(II) tetrahydroborate In tetrahydrofuran at 20℃; for 0.08h; regioselective reaction; |
The typical procedure for the regioselective 1,2-reduction of conjugated carbonyl compoundswith the Zn(BH4)2/Al2O3 system in THF
General procedure: In a round-bottomed flask (10 mL) equipped with a magnetic stirrer, a solution of benzylideneacetone(0.146 g, 1 mmol) in THF (3 mL) was prepared. To this solution, Zn(BH4)2(0.0.95 g, 1 mmol) and then neutral Al2O3 (0.101 g, 1 mmol) were added. The resulting mixturewas stirred at room temperature. The progress of the reaction was monitored by TLC(eluent, CCl4/Et2O: 5/2). After completion of the reaction within 60 min, distilled water (1mL) was added to the reaction mixture and this mixture was then stirred for an additional 5min. The mixture was extracted with CH2Cl2 (3×10 mL) and dried over anhydrous sodiumsulfate. Evaporation of the solvent and short column chromatography of the resulting crudematerial over silica gel (eluent, CCl4/Et2O: 5/2) afforded pure liquid 4-phenyl-3-buten-2-ol(0.140 g, 95 % yield, Table VIII, entry 3). |
| 92% |
With Co-doped ammonia borane In methanol at 20℃; for 0.0833333h; chemoselective reaction; |
|
| 91% |
With isopropanol at 120℃; for 4h; |
2.4. Catalytic reaction
General procedure: The MPV reaction of the biomass-derived compounds with 2-propanol was carried out in an oil-heated condition in a 15 ml Ace pressure tube (Synthware, Beijing). Typically, aldehydes (1.0 mmol), catalysts (0.1 g), and 2-propanol (10 mL) were added into the reactor, and then placed into the oil bath at stated temperature of 80-140 °C, followed bythe magnetic stirring for specific time at 600 rpm. After completion, the reaction tube was cooled to room temperature with cold water in a beaker. The reaction mixture was centrifuged and collected for analysis. Quantitative analysis of reactants and products on a standard sample using toluene as an internal standard on a GC (Shimadzu Nexis GC-2030) equipped with an HP-5 capillary column (30.0m×250mm×0.25 mm) and a flame ionization detector. Identification of products were observed using GC-MS (GCMS-QP2010 Ultra) equipped with HP-5MS capillary column (30.0m×250mm×0.25 mm). |
| 90% |
With benzoic acid sodium salt; sodium tetrahydridoborate; benzalacetophenone In water monomer at 20℃; for 0.5h; Sonication; Green chemistry; regioselective reaction; |
A typical Procedure for Competitive Reduction of aldehydes and ketones with NaBH4/PhCO2Na/H2O/U.S. system
General procedure: In a round-bottomed flask (10 ml) equipped with a magnetic stirrer, a solution of benzaldehyde (0.106 g, l mmol) and acetophenone (0.120 g, 1 mmol) in H2O (3 mL) was prepared. To this solution, NaBH4(0.047 g, 1.25 mmol) and PhCO2Na (0.138 g, 2 mmol) was added. The mixture was stirred and irradiated with ultrasound waves at room temperature for 60 minutes. TLC monitored the progress of reaction. After 60 min, the reaction mixture was quenched by addition of distilled water (5 ml) and this mixture was then stirred for an additional 1 min. The mixture was extracted with CH2Cl2(5×10 ml) and dried over anhydrous sodium sulfate.Evaporation of the solvent and short column chromatography of the resulting crude material over silicagel by eluent of Hexane/EtOAc: 9/1 affords the pure liquid benzyl alcohol (0.099 g, 92%) as a sole product of reduction and acetophenone as an intact material (table 2, entry 1) |
| 85% |
With iron(III) chloride; magnesium In water monomer; N,N-dimethyl-formamide at 20℃; for 2.5h; |
|
| 85% |
With (ethylenediamine)[1,2-bis(diphenylphosphino)ethane]ruthenium(II)(bisbenzoate); hydrogen In n-heptane at 80℃; Autoclave; Inert atmosphere; |
6 3,7-dimethylocta-2,6-dien-1-ol synthesis
3,7-Trimethyl-octa-2,6-dienal (as a 40/60 Z/E isomers mixture) (152 g, 1 mol.), heptane (304 g, 200 wt. %, technical grade) and (ethylenediamine)[1,2-bis(diphenylphosphino)ethane]ruthenium(bisbenzoate) (80.1 mg, 0.1 mmol, 0.01 mol.%) were loaded altogether in a 11 autoclave equipped with a mechanical stirring device. Sealed autoclave was then purged under stirring with nitrogen (3 times 5 bars) and hydrogen (3 times 5 bars) before being pressurized to 30 bars hydrogen. It was then heated to 80° C. and hydrogen pressure was maintained to 30 bars during all the reaction to afford desired product with 90% selectivity. Upon reaction completion (checked by both hydrogen consumption and GC), autoclave was cooled down to 25° C. It was then depressurized and purged with nitrogen (3 times 5 bars) and reaction mixture was then transferred to a round-bottomed flask and solvent was removed under vacuum. After initial flash distillation followed by further fractional distillation, pure 3,7-dimethyloct-2,6-dien-1-ol was obtained in 85% yield. |
| 80% |
With Zrβ-NO3-12.5 In isopropanol at 82℃; for 4h; |
|
| 74% |
With water monomer; 1,8-diazabicyclo[5.4.0]undec-7-ene; 4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl-4',4',5',5'-tetramethyl-1,3,2-dioxaborolane at 20℃; for 10h; Sealed tube; chemoselective reaction; |
|
|
With sodium hydroxide; sodium tetrahydridoborate; zinc 2-ethylhexanoate 1.) THF, reflux, 2.) 40 deg, 1 h; Yield given; Multistep reaction; |
|
|
With anhydrous sodium formate; isopropanol at 83℃; for 1h; |
|
|
With hydrogen at 120℃; Inert atmosphere; Autoclave; |
4
Example 4Selective Hydrogenation of Citral2 g of catalyst powder obtained in Example 1 was introduced into an autoclave of 200 ml in volume and then added with 130 ml of citral. After sealing the autoclave, nitrogen gas was repeatedly introduced into and discharged from the autoclave 3 times at a pressure of 1 MPa while stirring. Subsequently, the nitrogen gas was substituted with hydrogen gas at a pressure of 1.3 MPa and then heated up to 120° C. During the hydrogenation, samples were taken from a reaction vessel at regular intervals and analyzed by gas chromatography.The conversion rate of citral, the selectivity of nerol/geraniol generated on the basis of such a conversion rate, and the by-products are listed in Table 1. TABLE 1 Citral Selectivity of product (%) conversion Nerol/ Tetrahydro Unknown rate (%) geraniol Citronellal Citronellol geraniol substance 11.35 100.00 0.00 0.00 0.00 0.00 35.44 97.52 0.00 0.00 0.00 2.48 55.32 96.80 0.00 0.86 0.00 2.34 60.87 96.90 0.00 0.87 0.00 2.23 84.72 96.44 0.00 1.40 0.00 2.16 |
|
With hydrogen at 200℃; for 1h; Gas phase; chemoselective reaction; |
|
|
With gold supported on mesoporous ceria; hydrogen In water monomer at 100℃; for 2.5h; chemoselective reaction; |
|
| 52 %Chromat. |
With hydrogen; potassium hydroxide In isopropanol at 23℃; for 18h; autoclave; |
47
Examples 24-48; Catalytic hydrogenation of ketones or aldehydes using various invention's ruthenium complexes; The hydrogenation substrate (20 mmol), the base (as in Table 2), ο-propanol (10 ml), and the catalyst precursor RuCl2(L4) (0.01 mmol) were placed into a pressure reactor and stirred under ¾ (50 bar) at the given temperature for the given amount of time as indicated in Table 2. |
| 99 %Chromat. |
With formic acid; iron(II) tetrafluoroborate hexahydrate; tris(2-diphenylphosphinoethyl)phosphine In tetrahydrofuran at 60℃; for 2h; Schlenk technique; Inert atmosphere; |
2. Experimental
General procedure: Fe(BF4)2·6H2O (0.7 mg; 0.002 mmol) and tris[2-(diphenyl-phosphino)-ethyl]phosphine [P(CH2CH2PPh2)3; tetraphos] (1.4 mg; 0.002 mmol) are placed in a Schlenk-tube under argon atmosphere. 1 mL dry tetrahydrofurane is added and the purple solution is stirred for 2 min. Cinnamaldehyde (63 μL; 0.5 mmol) and 100 μL n-hexadecane as an internal GC-standard are injected and a sample is taken for GC-analysis. The solution is heated to 60 °C and the reaction starts by addition of 1.1 equiv formic acid (22 μL; 0.55 mmol). After 2 h, a second sample is taken for GC-analysis and conversion and yield are determined by comparison with authentic samples. For the isolation, the reaction is scaled up by a factor of 20. When the reaction is completed, the reaction solution is diluted with a mixture of n-hexane and ethyl acetate (3:1), filtered through a plug of silica and the solvent removed in vacuum. |
| 52 %Chromat. |
With (N,N'-bis(2-(tert-butylthio-kS)benzylidene)-1,2-ethanediamino-kN,kN')dichlororuthenium(II); hydrogen; potassium hydroxide In isopropanol at 23℃; for 18h; Inert atmosphere; Autoclave; |
47
General procedure: The hydrogenation substrate (20 mmol), the base (as in Table 2), iso-propanol (10 ml), and the catalyst precursor RuCl2(L4) (0.01 mmol) were placed into a pressure reactor and stirred under H2 (50 bar) at the given temperature for the given amount of time as indicated in Table 2. |
|
With [(ethylenediamine)(9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene)Ru(OCOtBu)2]; hydrogen; benzoic acid at 100℃; for 5h; Autoclave; |
|
|
With hydrogen In hexane at 60℃; for 18h; Autoclave; chemoselective reaction; |
|
|
With hydrogen In isopropanol at 90℃; |
|
|
With hydrogen In ethanol; butan-1-ol at 80℃; for 1h; chemoselective reaction; |
|
|
With hydrogen In hexane at 60℃; for 18h; Autoclave; chemoselective reaction; |
|
| > 99 %Chromat. |
With C16H33Cl2CoN5P2; hydrogen; sodium tertiary butoxide In tert-Amyl alcohol at 20℃; for 24h; Autoclave; |
|
| 87.6 %Chromat. |
With porous zirconium-phytic acid hybrid In isopropanol at 100℃; for 6h; |
|
|
Multi-step reaction with 2 steps
1: [bis(2,6-diisopropylaniline)acenaphthene]Fe(η6-toluene) / neat (no solvent) / 70 °C / Glovebox; Schlenk technique
2: N,N,N-tributylbutan-1-aminium fluoride / tetrahydrofuran / 3 h / 0 - 20 °C |
|
| 73 %Spectr. |
With C16H26AlNO; isopropanol for 0.25h; Reflux; Inert atmosphere; Schlenk technique; |
4.3 Typical procedure employed for MPV reactions
General procedure: A 25mL Schlenk flask was charged with aluminum complex (0.2mmol) and the carbonyl compound (4.0mmol) was added, followed by the addition of 2-propanol (0.37mL, 4.8mmol). The reaction mixture was then refluxed for 15min, and the yield was determined by 1H NMR spectroscopic studies based on the integration in the methylene and the CHO region of the benzyl group. |
|
With hydrogen In ethanol; water monomer at 99.84℃; for 4h; Autoclave; Green chemistry; chemoselective reaction; |
|
| 99 %Spectr. |
With [Fe(PNPMe-iPr)(CO)(H)(Br)]; hydrogen; 1,8-diazabicyclo[5.4.0]undec-7-ene In ethanol at 40℃; for 16h; chemoselective reaction; |
|
|
With isopropanol at 120℃; for 24h; chemoselective reaction; |
2.3.2. MPV-reduction
General procedure: Before reaction, each catalyst was dried at 200 °C to remove residual solvent molecules; catalytic reactions were carried out in 10 ml glass crimp cap vials loaded with 20-30 mg catalyst and a magnetic stirring bar. A solution of the substrate in 3.3 mL isopropanol (IPA) was added; n-tetradecane was added as internal standard. For each catalyst, a substrate to Zr ratio of 7.8 was used as to compare the activity of each catalyst. After introduction of the reaction mixture, the vials were placed in an aluminium heating block (at 120 °C) and stirred. Reaction samples were filtered through a 0.2 μm filter and analysed with gas chromatography (Shimadzu 2010 GC, CP-Sil 8, FID detector). Reaction products were identified using GC-MS. Reactions were performed in duplo, and the results shown are averaged. |
|
With hydrogen In neat (no solvent) at 110℃; for 12h; chemoselective reaction; |
|
|
With isopropanol at 160℃; for 48h; Inert atmosphere; Sealed tube; |
|
|
With C39H51AlN6; isopropanol In toluene at 110℃; for 2h; Inert atmosphere; Schlenk technique; |
2.5. Typical procedure employed for the Meerwein-Ponndorf-Verley(MPV) reduction
General procedure: To a solution of catalyst (0.4 mmol) in toluene (10 mL) wasadded 2-propanol (8.0 mmol), followed by the addition of the aldehyde(4.0 mmol). The reaction mixture was then refluxed for therequired reaction time under an atmosphere of nitrogen. The reactionwas then cooled to room temperature and the conversionyield was determined by 1H NMR spectroscopic study based onthe integration in the methylene and the CHO region. |
| 94 %Chromat. |
With isopropanol at 120℃; for 6h; |
|
| 70.2 %Spectr. |
With C40H86Li2Mg2N4O2; isopropanol In neat (no solvent) for 4h; Inert atmosphere; Schlenk technique; Reflux; |
|
| 89 %Spectr. |
With [{(C5H10N)C(NCy)2}AlMe(μ-OMe)]2; isopropanol In toluene at 110℃; for 8h; Reflux; Inert atmosphere; Schlenk technique; |
2.6 General procedure employed for the MPV reaction
General procedure: To a solution of catalyst in toluene was added aldehyde or ketone, followed by the addition of 2-propanol. The reaction mixture was then refluxed for the required reaction time under an atmosphere of nitrogen. The reaction was then cooled to room temperature and the conversion yield was determined by 1H NMR spectroscopic study based on the integration in the methylene and the CHO region. |
|
With hydrogen In isopropanol at 149.84℃; for 0.75h; Autoclave; chemoselective reaction; |
|
|
Multi-step reaction with 2 steps
1: 3-bromo-2-(tert-butyl)-4H-5-(2-pyridyl)-1,2,4,3-triazaphospholene / acetonitrile / 16 h / 20 °C / Glovebox
2: potassium hydroxide / dichloromethane / 0.5 h / 20 °C / Glovebox |
|
|
With nanocrystalline copper oxide supported on magnesia; cyclohexanol at 180℃; Autoclave; chemoselective reaction; |
|
| 97 %Chromat. |
With Ru(NNS<SUP>Me</SUP>)(PPh<SUB>3</SUB>)Cl2; potassium-t-butoxide; hydrogen In tetrahydrofuran; isopropanol at 80℃; for 16h; Inert atmosphere; |
17a Example 17: hydrogenation of different aldehydes/ketones
General procedure: 4 mL glass reaction vials and stirring bars were dried overnight at 1 10°C, closed with PTFE/rubber septa, placed in a multiple reactor inlet suitable for a pressure vessel, and brought under argon atmosphere by three vacuum-argon cycles. With a syringe the reaction vessels were charged with the catalyst as stock solution in /PrOH (1 mL, 0.0005 mol/L, 0.05 mol%), followed by a solution of the compound A, B or C in /PrOH (1 mL, 1 mol/L, 1 mmol). After that a solution of freshly sublimed the base in THF (12.5 μ, 1 mol/L, 0.0125 mmol, 1 .25 mol%) was added with a Hamilton syringe. The reaction mixtures were transferred to an argon-filled pressure vessel, which was im- mediately flushed with three nitrogen and three hydrogen cycles, then pressurized to 30 bar hydrogen, heated to 80°C and stirred for 16h. After that the pressure vessel was cooled down to room temperature and depressurized. The reaction mixtures were filtered over silica and rinsed with ethanol (2 mL). The products are determined based on GC analysis retention time. The given values [%] are related to GC area%. The results are summarized in the following tables 4a, 4b and 4c. |
|
With Co3O4/mesoporous carbon In isopropanol at 120℃; for 48h; Inert atmosphere; |
Transfer hvdroqenation of substrates
For a typical run, 1 mmol of substrates, 10 mL of 2-propanol, 50 mg of Co3O4 MC and a magnet bar were placed in a glass vial (20 mL). The vial was flushed with argon and then tightly closed. The experiment was performed at 120 °C under magnetic stirring of 800 rpm in a stainless steel heating block. A small volume of sample (-0.1 mL) was periodically withdrawn and analyzed by GC-MS. 1 ,6- hexandiol was chosen as internal standard for FAL and HMF system, while n- decane was used as internal standard for cinnamaldehyde and citral. When increasing reaction temperature to 140 °C and 160 °C, the experiments were carried out in a stainless steel autoclave reactor with volume of 20 mL. The rest of experimental conditions remained the same. |
|
With C24H37IrN2O2P2; C44H68Ir2N2O4P4; hydrogen In tetrahydrofuran at 21.84℃; for 5h; chemoselective reaction; |
|
|
With Ru(H)<SUB>2</SUB>(CO)(triphos); hydrogen In neat (no solvent) at 100℃; for 16h; chemoselective reaction; |
|
| 90 %Chromat. |
With isopropanol at 140℃; for 5h; |
|
|
With isopropanol at 160℃; for 6h; Autoclave; Inert atmosphere; Sealed tube; |
|
| 76 %Spectr. |
With C20H39BrFeN3OP2; water monomer; anhydrous sodium formate at 80℃; for 1h; chemoselective reaction; |
General procedure for the catalytic transferhydrogenation of aldehydes
General procedure: In a typical experiment, a vial containing a magnetic stirringbar was charged with catalyst 1 and the substrate(2.0 mmol) inside a glovebox. The vial was sealed with aseptum screw cap, taken out from the glovebox and asolution of sodium formate in degassed water (1.0 cm3,2.5 M) was added through the septum. The reaction mixturewas stirred at 80 C for the specified time after whichit was quickly cooled to room temperature and the reactionwas quenched by exposure to air. A sample was taken fromthe organic phase, diluted in CDCl3, and analyzed by NMRspectroscopy. For the isolation of the reaction products,1 cm3 diethyl ether was added and the phases were separated.The aqueous phase was washed with diethyl etherand the combined extracts were filtered over a short plug ofsilica to remove the catalyst. The solution was dried overMgSO4 and the solvent removed under reduced pressure. |
|
With bis(tricyclohexylphosphine)-3-phenyl-1H-inden-1-ylidene ruthenium(II)dichloride; anhydrous sodium formate; n-dodecyltrimethylammonium chloride In water monomer at 85℃; Schlenk technique; |
|
|
With activated carbon supported CoOx-shell/Co-core structured nanoparticle calcined at 500°C In isopropanol at 120℃; for 13h; Inert atmosphere; |
|
|
With potassium-t-butoxide; C55H44ClFeN2OP2(1+)*BF4(1-); isopropanol at 25℃; for 0.05h; Inert atmosphere; chemoselective reaction; |
|
| 91 %Spectr. |
With methanol; borane-ammonia complex at 20℃; for 0.5h; Inert atmosphere; Schlenk technique; Glovebox; Green chemistry; |
|
|
With naphthalene; MOF-808 In isopropanol at 82℃; for 1h; |
Catalytic test and product analysis
General procedure: The catalytic activity of Zr-MOFs in the transfer hydrogenation of flavoring compounds was evaluated at moderate reaction conditions. Before use, Zr and Hf-MOFs were dried in an oven at 100°C for 12 h. In a typical procedure, a known amount of dried catalyst (9.5 mol% on the metal basis) of the dried catalyst, 2.6 mmol of carbonyl substrate, 0.05 g of naphthalene as an internal slandered, and 20 ml of isopropanol were added into a two-neck round bottom flask of 50 mL capacity. The reaction mixture in RBF equipped with a rubber septum and reflux condenser was heated at the boiling point of isopropanol for the desired time. The catalyst was separated by centrifugation and washed thoroughly with methanol and dried at 100°C. The filtrate was subjected to quantitative analysis using gas chromatography (GC, FID detector, and DB-624 column). Substrate conversion and the yield of products were determined using a single point internal standard calibration method. |
|
With Zr(OH)4/CoFe2O4 core-shell magnetic nanoparticles; alcohol at 160℃; for 4h; Autoclave; |
|
|
With hydrogen; C27H41IrN3P In 2-methyltetrahydrofuran at 25℃; for 24h; Inert atmosphere; Glovebox; chemoselective reaction; |
|
|
With Hf-MOF-808 catalyst In isopropanol at 82℃; for 2h; |
Table 7 below shows an experiment for hydrogenation of the M-MOF-808 catalyst when the substrate is α,β-unsaturated carbonyl. The reaction condition of Table 7 below was the same as that of Table 6, except that the reaction temperature was 82° C. In the table below, the type of the central metal of MOF-808 was set to Zr or Hf. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping