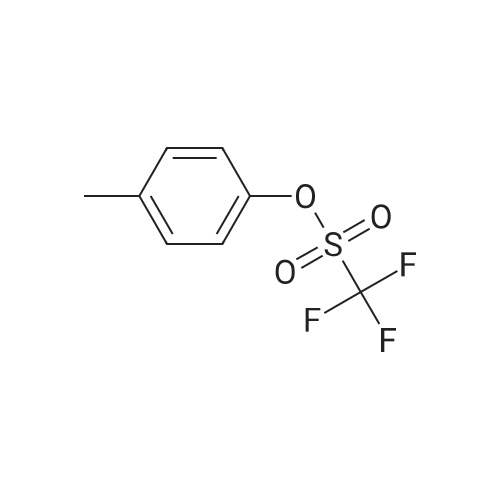
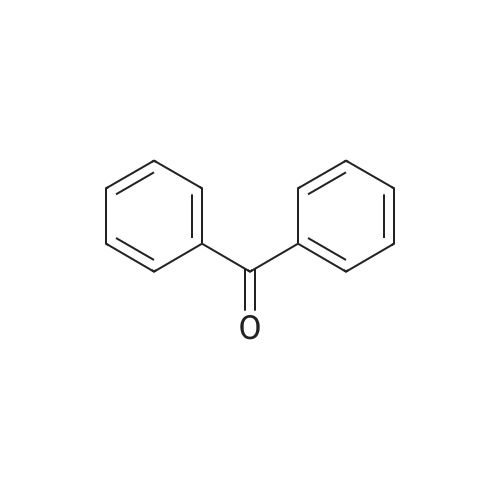
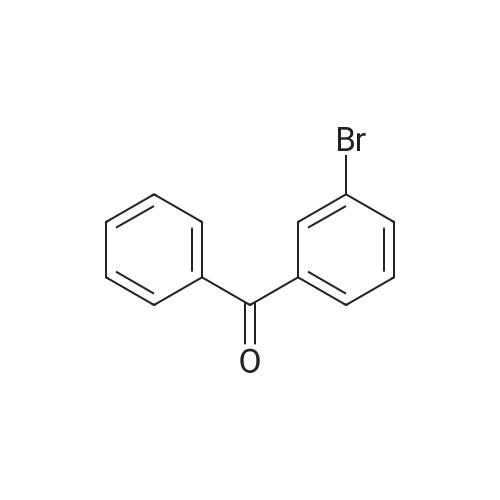
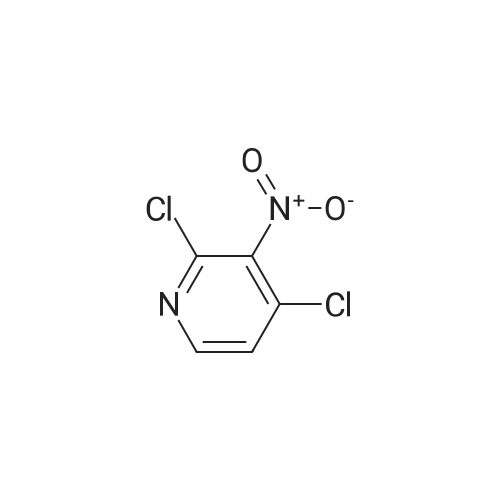
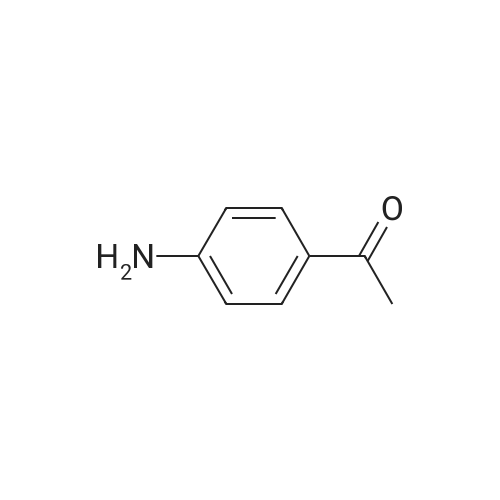
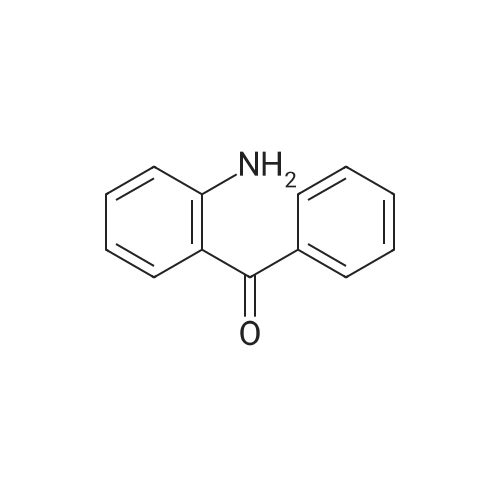
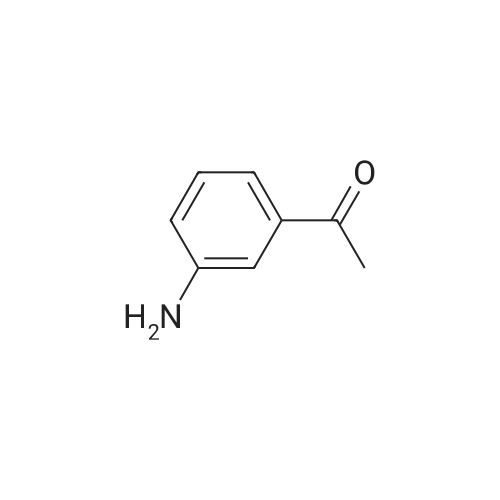
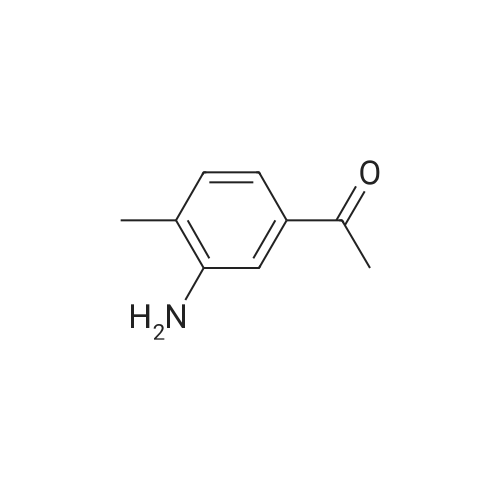
| 97% |
With sodium tetrahydridoborate; NiCl2·6H2O In lithium hydroxide monohydrate at 20℃; for 3h; Inert atmosphere; Green chemistry; |
|
| 96% |
With trichlorosilane; N-ethyl-N,N-diisopropylamine In dichloromethane at 25℃; for 0.0833333h; Flow reactor; |
General procedure for the continuous-flow reaction using a0.5 mL PTFE reactor
General procedure: Syringe A was filled with a solution ofHSiCl3 (2.4 mmol) in dry CH2Cl2 (1.5 mL). Syringe B wasloaded with a solution of the nitro compound (0.6 mmol) andHünig’s base (3.6 mmol) in dry CH2Cl2 (1.5 mL). Syringes Aand B were connected to a syringe pump and the reagents werepumped into the microreactor at the indicated flow rate(mL/min) at room temperature. The outcome of the reactor wascollected in a flask containing a 10% NaOH solution. Fivereactor volumes were collected. CH2Cl2 was removed in vacuoand the aqueous layer was extracted three times with ethylacetate. The combined organic layers were washed with brine,dried with Na2SO4 and concentrated in vacuo. 1H NMR spectroscopy of the crude was used to calculate the reaction conversion; in case of a full conversion of the starting material nofurther purification was required |
| 95% |
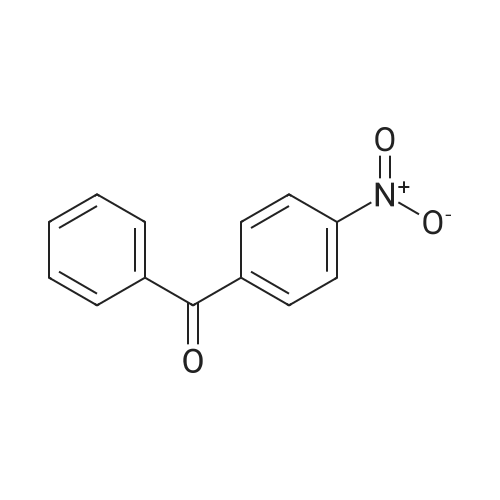
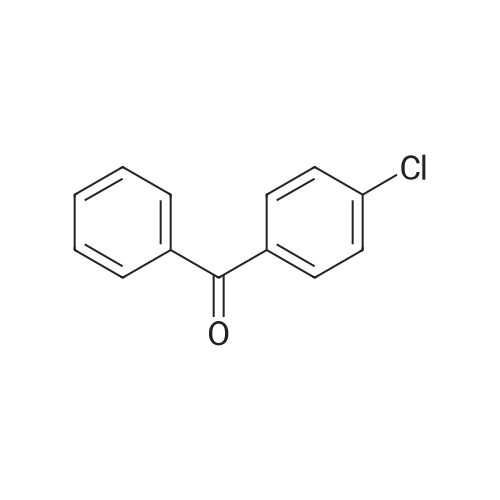
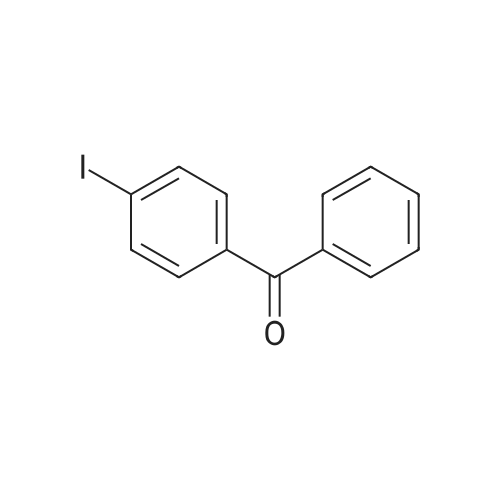
Stage #1: (4-nitrophenyl)(phenyl)methanone With hydrogen bromide; hypophosphorous acid; glacial acetic acid; sodium iodide In lithium hydroxide monohydrate at 115℃; for 3h; Inert atmosphere;
Stage #2: With sodium hydroxide In lithium hydroxide monohydrate Inert atmosphere; |
|
| 95% |
Stage #1: (4-nitrophenyl)(phenyl)methanone With N-phenylhydrazine monohydrochloride at 65℃;
Stage #2: With 5-aminotetrazole Reflux; |
8 synthesis of p-aminobenzophenone
A ketoprofen intermediate for the synthesis of p-aminobenzophenone, comprising the steps of:A, in a reaction vessel equipped with a stirrer, a reflux condenser, and a dropping funnel, 700 ml of a phenylhydrazine hydrochloride solution, 0.61 mol of p-nitrobenzophenone was added and the stirring speed was controlled at 170 rpm, Temperature to 65 , reflux 3h;B, dropping 43% 5-aminotetrazole solution 300ml, dropping time control in 3h, after adding the continued reflux 90min, 3.3kPa vacuum distillation 2h;C, reduce the temperature of the solution to 10 , filter, ammonium nitrate solution, add it to 1.5L mass fraction of 33% oxalic acid solution, raise the temperature of the solution to 70 , keep 60min, filter, add the filtrate 300ml mass fraction 27% potassium sulfite solution, precipitation of solid, reduce the temperature of the solution to 20 , filtration, the mass fraction of 15% sodium bromide solution washing, anhydrous calcium chloride dehydration agent dehydration, p-amino benzophenone 113.56g, yield 94%. The mass fraction of the sulfite solution in Example 1 is adjusted to the same as the remaining ratio of raw materials and preparation conditions in Example to obtain a reaction yield Seen from Example 8, the yield of the reaction mass is proportional to the fraction of the solution with sulfite, potassium sulfite solution, the mass fraction of low yield great influence on the reaction, considering the cost, the sulfite solution is preferably a mass fraction of 25-29%. |
| 95% |
With hydrogen In 2-methyltetrahydrofuran; lithium hydroxide monohydrate at 40℃; for 24h; chemoselective reaction; |
|
| 93% |
With 2,3-dimethyl-2,3-butane diol; MoO<SUB>2</SUB>Cl<SUB>2</SUB>(DMF)<SUB>2</SUB> In toluene at 150℃; for 0.333333h; Microwave irradiation; chemoselective reaction; |
|
| 93% |
With trichlorosilane; N-ethyl-N,N-diisopropylamine In acetonitrile at 0 - 20℃; for 18h; Inert atmosphere; chemoselective reaction; |
|
| 93% |
Stage #1: (4-nitrophenyl)(phenyl)methanone With hydrogenchloride; 1,1,1,3',3',3'-hexafluoro-propanol; iron(0) In lithium hydroxide monohydrate at 20℃; for 0.5h;
Stage #2: With Sodium hydrogenocarbonate In lithium hydroxide monohydrate chemoselective reaction; |
2. General Procedure for the Reduction of Nitro Compounds
General procedure: The nitro compound (1 equiv), HFIP (10 equiv), Fe powder (5 equiv) were mixed in a tube. Then 2 N HCl aqueous solutions was added to the reaction mixture. After stirring at room temperature for 30 min, the reaction mixture was neutralized with sat. NaHCO3 (aq.) and extracted with EtOAc three times. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was then purified by column chromatography on silica gel to furnish the desired amine product. |
| 92% |
With anhydrous ammonium formate; zinc In methanol at 20℃; for 0.166667h; |
|
| 92% |
With hydrogen In ethanol at 60℃; for 10h; |
13 Preparation of 4-aminobenzophenone
Nano-porous metal cesium catalyst (3.2 mg, 0.03 mmol) Ethanol (lmL) and 4-nitrobenzophenone (68.17mg, 0.3mmol) were added to the reaction kettle, hydrogen (10 bar) was added, heated and stirred, the reaction temperature was controlled at 60 ° C, The reaction time was controlled at 10 h, column chromatography (baby gel, 200-300 mesh; developing solvent, petroleum ether: ethyl acetate = 10: 1) to give 54.4 mg of 4-aminobenzophenone in 92% yield. |
| 92% |
With hydrogen In toluene at 140℃; for 48h; chemoselective reaction; |
|
| 91% |
With sodium hydroxide; carbon monoxide In lithium hydroxide monohydrate; toluene at 30℃; for 1.08333h; specific rate of reduction under biphasic condition; |
|
| 91% |
With iron(0); glacial acetic acid In ethanol for 2h; Heating; |
|
| 91% |
With triethylamine In lithium hydroxide monohydrate at 80℃; for 6h; Inert atmosphere; Green chemistry; chemoselective reaction; |
|
| 90% |
With potassium hydroxide; isopropanol for 3h; Heating; |
|
| 90% |
With formic acid; nickel In methanol at 20℃; for 0.333333h; |
|
| 90% |
With formic acid at 140℃; for 0.133333h; Microwave irradiation; |
|
| 89% |
With nickel oxide; aluminum(III) oxide; potassium hydroxide; isopropanol In nitrobenzene for 1h; Heating; |
|
| 89% |
With potassium hydroxide; isopropanol for 1h; Heating; |
|
| 89% |
With sodium tetrahydridoborate; lithium hydroxide monohydrate at 20℃; for 1h; |
2.3. Heterogeneous reduction of nitroarenes catalyzed by the CoFe2O4Pd/AC nanocomposite
Reduction of nitroarenes was carried out in a 10 mL round-bottomflask equipped with a magnetic stirrer. First, 6 mg of the CoFe2O4Pd/AC was dispersed in 3 mL deionized water. Then, 1 mmol of the nitroarenewas added to the flask. With a continuous stirring at room temperature,1.5 mmol of NaBH4 in 4 mL deionized water was slowly addedto the reaction vessel during stirring. After appropriate time, thecompletion of the reaction was monitored by thin-layer chromatography(TLC). The catalyst was separated by using an external magnet from thereaction mixture. The mixture was decanted and extracted by ethyl acetateand dried over anhydrous Mg2SO4. The isolated product was obtainedby evaporating the ethyl acetate and subjected to characterizationby IR spectroscopy. |
| 85% |
With sodium hydroxide; baker's yeast In methanol; lithium hydroxide monohydrate at 70 - 80℃; for 5h; |
|
| 84% |
With stainless steel; lithium hydroxide monohydrate; chromium; zirconium oxide for 3h; |
9 Example 9 Synthesis of 4-Aminobenzophenone Through Hydrogenation Reaction of 4-Nitrobenzophenone
Example 9 Synthesis of 4-Aminobenzophenone Through Hydrogenation Reaction of 4-Nitrobenzophenone [0112] 91.1 mg (0.50 mmol) of 4-nitrobenzophenone (7) and 270 μL (15 mmol) of distilled water were placed in a planetary ball mill vessel (12 mL), in which 50 pieces of balls made of zirconia (diameter: 5 to 6 mm) and 78 mg (1.5 mmol) of chromium powder were placed, and the equipment was then closed and rotated for 3 hours at 800 rpm (reversed every 30 minutes) for agitation. After a lapse of 3 hours, 10 mL of ethyl acetate was added to the planetary ball mill vessel to provide a solution containing the reaction mixture, which was then filtered with Celite. The operation was repeated 5 times to provide a filtrate, which was then concentrated, and then 1H NMR confirmed that 4-aminobenzophenone (8) was obtained. The yield was 84%. The reaction is expressed by the following scheme. |
| 83% |
With tetrahydroxydiborane; 5%-palladium/activated carbon; lithium hydroxide monohydrate In acetonitrile at 50℃; for 24h; chemoselective reaction; |
4.2 Typical procedure for reduction of nitrobenzene
General procedure: Nitrobenzene (0.6mmol), 5wt% Pd/C (0.5mmol %, 0.003mmol), H2O (10 equiv, 6.0mmol), B2(OH)4 (3.3 equiv, 2.0mmol), and CH3CN (1.0mL) were added in a 10mL tube. The reaction mixture was stirred at 50°C for 24h. When the reaction was complete monitored by TLC, the mixture was cooled to room temperature. Water (5mL) was added, and extracted with EtOAc (3×5mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give aniline 2a (55mg, 99%). |
| 82% |
With sodium tetrahydridoborate In tetrahydrofuran; lithium hydroxide monohydrate at 20℃; for 4h; Inert atmosphere; Green chemistry; chemoselective reaction; |
|
| 81% |
With tetrahydroxydiborane; palladium on activated charcoal; lithium hydroxide monohydrate In acetonitrile at 50℃; for 24h; Inert atmosphere; |
16; 41; 52 Example 16Synthesis of 4-aminobenzophenone
4-nitrobenzophenone (0.6mmol, 99.1mg), water (6mmol, 108.0mg), Pd/C (0.03mmol, 6.4mg) and tetrahydroxydiboron (1.98mmol, 177.5mg), acetonitrile (1mL ), under the protection of nitrogen, react at 50C for 24h, monitor the reaction by TLC, add 10mL water, extract with ethyl acetate (10mL×3), combine the organic phases, dry with anhydrous sodium sulfate, filter, concentrate under reduced pressure, and separate by column chromatography (V Petroleum ether: V ethyl acetate = 3:1) to obtain 95.9 mg of a yellow solid, that is, to obtain the target compound with a yield of 81%. |
| 72% |
With titanium(IV) tetrachloride; diisobutyl telluride In dichloromethane for 0.5h; Ambient temperature; |
|
| 72% |
With Zr12(μ3-O)5[(μ3-O)CoCl]8[(μ2-O)2(μ3-O)CoCl]3Li3(triphenyldicarboxylate)9; hydrogen; sodium triethylborohydride In toluene at 110℃; for 42h; Inert atmosphere; Schlenk technique; |
|
| 71% |
With aluminum(III) oxide; dicobalt octacarbonyl In hexane |
|
| 71% |
With tetrahydroxydiborane; copper (II) acetate In acetonitrile at 80℃; for 24h; Schlenk technique; chemoselective reaction; |
4.1.2. Typical procedure for the synthesis of 8-Aminoquinoline (4a)
General procedure: A 20 mL Schlenk tube was charged with 8-nitroquinoline (1k; 87 mg, 0.5 mmol), Cu(OAc)2 (4.5 mg, 0.025 mmol), B2(OH)4(135 mg, 1.5 mmol), and MeCN (2.0 mL). The mixture was stirred at 80 °C for 24 h, then cooled to room temperature and concentrated under reduced pressure. Similar workup to 2a gave a brown solid (4a: 63 mg, 87% yield). |
| 63% |
With iron sulphate heptahydrate; Aluminum Chloride; zinc In ethanol; lithium hydroxide monohydrate at 50℃; for 2h; |
|
|
With hydrogenchloride; ethanol; stannous chloride |
|
|
With 1,4-dioxane; platinum Hydrogenation; |
|
|
With ethanol; nickel Hydrogenation; |
|
|
With tin; glacial acetic acid |
|
|
With reducing agent |
|
|
With hydrogenchloride; iron(0); ammonia hydrochloride In ethanol at 55 - 65℃; |
|
| 100 %Chromat. |
With titanium dioxide In ethanol for 1.25h; Irradiation; Green chemistry; chemoselective reaction; |
Photocatalytic synthesis of aromatic amines
General procedure: Photocatalytic synthesis of aromatic amines. Photocatalytic reactions were carried out in a round bottomed Pyrex flask and irradiated using four high power blue light LEDs 3W lamp or by solar light under magnetic stirring at room temperature. Reaction conditions with solar light: the reduction of the aromatic nitro compounds (0.02 mmol) was carried out in the presence of TiO2-P25 (0.01 g) in EtOH (4 mL) with irradiation for 1-4 h. |
| 100 %Chromat. |
With oxalic acid; titanium(IV) dioxide; β‐cyclodextrin In lithium hydroxide monohydrate for 2h; Inert atmosphere; Irradiation; Green chemistry; |
|
| 93 %Chromat. |
With cadmium sulphide In isopropanol for 1.16667h; Inert atmosphere; Sonication; Irradiation; |
Experimental
General procedure: As optimized reaction conditions, a solution of nitroaromaticcompounds (0.01 M) in an appropriatesolvent and 20 mg of CdS-NP were sonicated andslowly purged with N2 for 5 min. Then, the reactionvessel was sealed up with a rubber stopper and themixture was stirred magnetically and irradiated with blue LED (4 × 1 W, λ ≥ 420 nm, intensity: 80 lumen)or sunlight (of daily ambient temperature andsunlight intensity range of 80-100 × 103 lux).The reaction conversion was monitored by thinlayer chromatography (TLC). After completing thereaction, the mixture was centrifuged and supernatantwas removed and analyzed on a GC Alientgas chromatograph (Nonpolar CP-Sil 8 column (30m × 0.32 mm), Varian Chrompack (Middelburg, TheNetherlands). |
|
With ammonia; hydrogen In methanol at 90℃; for 38h; Autoclave; |
|
| 93 %Chromat. |
With hydrogen; triethylamine In ethanol; lithium hydroxide monohydrate at 110℃; for 22h; Autoclave; chemoselective reaction; |
|
| 95 %Chromat. |
With cadmium sulfide loaded on silica-coated Fe3O4 nanoparticles In isopropanol for 20h; Irradiation; Inert atmosphere; |
2.3 Photocatalytic activity
General procedure: The Fe3O4/SiO2/CdS (S2) with average amount of CdS has been chosen as a photocatalyst for the photocatalytic reduction of nitro compounds under the blue LED irradiation. In a 10mL flask, 5ml of 0.01M nitro compounds solution and 0.02g photocatalyst were charged. Then the flask was charged with pure argon. The resulting mixture was stirred for 20h under LED irradiation. After this time, the catalyst was simply separated by employing an external magnetic field and the remaining solution was analyzed using thin-layered chromatography (TLC) and Varian gas chromatograph (CP-3800). The conversion of nitro substrate, yield of amine, and selectivity for amine were defined as follows: Conversion (%) = (C0-Cnitro)/C0×100 Yield (%) = Camine/C0×100 Selectivity (%) =Camine/(C0-Cnitro)×100Where C0 is the initial concentration of nitro compound and Cnitro and Camine are the concentration of the nitro substrate and the corresponding amine respectively, after the photocatalytic reaction. |
| 93 %Chromat. |
With hydrogen; triethylamine In ethanol; lithium hydroxide monohydrate at 110℃; for 22h; Autoclave; |
3.1.1; 3.1.9 General Procedure for the Preparation of substituted Anilines from Nitroarenes
General procedure: In a 4 ml_ reaction glass vial fitted with a septum cap containing a magnetic stirring bar, Co-Co3O4Chit-700 (10 mg, 3.4 mol% Co), the nitroarenes (0.5 mmol, 1 .0 equiv.) and triethylamine (35 μΙ_, 0.25 mmol, 0.5 equiv.) were added to a solvent mixture of EtOH/H20 (3/1 , 2 ml_). The reaction vial was then placed into a 300 ml_ autoclave, flashed with hydrogen five times and finally pressurized to 40 bar. The reaction mixture was stirred for appropriate time at 1 10 °C. After cooling the reaction mixture to room temperature, the autoclave was slowly depressurized. The crude reaction mixture was filtered through a pipette fitted with a cotton bed and the solvent was evaporated under reduced pressure. The crude products were purified by passing through a silica plug (eluent: ethyl acetate) to give pure aniline derivatives after removal of solvent. The following compounds may be prepared from the respective nitroarenes using the catalyst of the invention: |
| > 99 %Chromat. |
With tris(2,2′-bipyridine)ruthenium(II) hexafluorophosphate; 3C93H72N8O4P4(4-)*6Co(2+); quinhydrone; L-ascorbic acid In lithium hydroxide monohydrate; acetonitrile at 24.84℃; for 12h; Inert atmosphere; Irradiation; |
4.3 General procedure for selective hydrogenation of nitroarenes to anilines
General procedure: In a typical experiment, selective hydrogenation of nitroarenes to anilines was made in a 20mL flask. Varying amounts of the p-nitroacetophenone, the catalyst, Ru(bpy)32+ and ascorbic acid in 1:1 CH3CN/H2O were added to obtain a total volume of 5.0mL. The flask was sealed with a septum, degassed by bubbling argon for 15min under atmospheric pressure at room temperature. The pH of this solution was adjusted to a specific pH by adding H2SO4 or NaOH and measured with a pH meter. After that, the samples were irradiated by a 500W Xenon Lamp, the reaction temperature was 298K by using a water filter to absorb heat. The generated photoproduct of selective hydrogenation of nitroarenes evolution was characterized by GC-MS analysis. |
|
With hydrogen In methanol; lithium hydroxide monohydrate at 90℃; for 5h; |
|
| 90 %Chromat. |
With sodium tetrahydridoborate In ethanol; lithium hydroxide monohydrate at 20℃; for 1.5h; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping